
Molecular Transduction of Mechanical Signals: Simulation Study of Force-Induced Conformational Change
Aurore Zyto, Roger D. Kamm & Bruce Tidor
Cells can sense and respond to their mechanical environment, including forces applied externally, or cell-generated. Biological behaviors regulated by mechanical signals include growth, differentiation, apoptosis, motility, and gene expression [1]. Experimental observations have demonstrated the mechanical nature of these phenomena, but the process by which these signals result in cellular change is largely unknown. One hypothesis is that key protein molecules act as "mechanosensors" and "mechanotransducers"; the combined effect of these functions is to convert a mechanical signal into a chemical one that more easily connects to cellular biochemistry and downstream effectors. One mechanism for achieving mechanotransduction is through force-driven conformational change, and a more generalized mechanism is force-modulated binding affinity [2]. In this work, we apply computational modeling to investigate key features of molecular events involved in mechanotransduction. The computational studies correspond to biological systems that can be studied with single-molecule experiments. Our focus is on elucidating biomolecule structure-function relationships and the processes by which they change in response to mechanical force. Progress in development of general approaches to study force-induced changes in the structural properties and associated biochemical activity of proteins is illustrated.
Method Development
To develop a theoretical framework to validate our methodology, we designed a simple mechanical system. A triangular shaped "ligand" is allowed to fit into a complementary grove in a second block. We analytically compute both the equilibrium position and associated "binding energy" as functions of the applied force on the "protein" block.
We identified two biological systems of interest. Four-helix bundles are a simple, common, motif of protein structure of regular toppology. Several focal adhesion proteins, including some potential "mechanosensors", are made up of those domains. We chose to focus on a member of this class - FRAP binding domain, seeking to extract out general principles of force sensing and transduction at the molecular level, allowing for expansion to other protein systems. The second system of interest consists of zinc fingers domain binding to DNA. This system is suitable for both computer analysis and experimental validation using single-molecule techniques [3].
We perform Molecular Dynamics simulations under force application, to assess the effects of force on protein conformational states. Implicit solvent calculations enable us to determine force directions of interest. We are also working on using explicit solvent models, to more accurately capture force effects. Normal modes analysis helps us to identify natural motions of the biomolecule, which are thought to be important for the force response characteristics. Finally, we are developing a technique, based on thermodynamics theory, to compute changes in free energy as a function of applied force, to study force-induced changes in binding affinity.
Preliminary Results
Design of a simple mechanical system helps understanding the possible effects of force on binding affinity. With our simple model, we observed three regimes of force application, due to a pre-compressive force that was applied on the top spring. In the first one, for moderate forces, the bound state does not change conformation. However the unbound state is altered, leading to overall loss of binding affinity. In the second case, both the bound and unbound states adopt deformed conformations due to force application, resulting in a larger decrease in binding affinity. Finally, binding is completely disrupted for very high forces. Hence accurate modeling of the unbound state, and its modifications due to response to force, is critical to studying force impact on binding characteristics.
Proteins can respond very differently to a mechanical stimulus, depending not only on its amplitude, but also on its mode of application. We addressed the following question: if we pull only at "extremities" of a four-helix bundle, what are the directions that have the greatest impact on structural and binding characteristics? We selected four residues located at the top of the bundle, and performed pulling simulations with constant force on all pairs of residues from this set. The protein has different responses: in some cases, its structure is deformed to a new conformational state, that is stable for the length of the simulation, even at high forces. For other pulling directions, the protein starts to unfold, for the same mode of forcing.
Normal mode analysis is a first step towards understanding global motions of the protein of interest, and the correlation between force application and displacement along those easy motions.
Research Support
This work is supported by the National Institutes of Health.
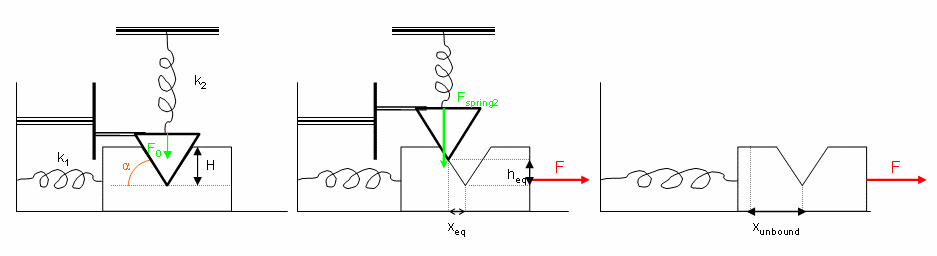
Designed mechanical system as a theoretical tool for computing binding free energy as a function of force.
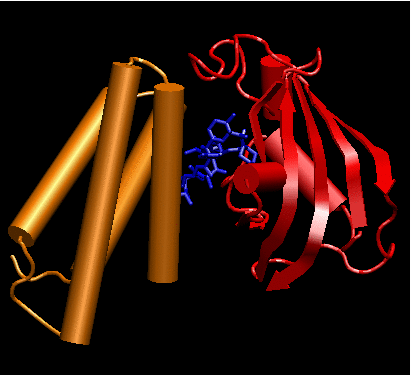
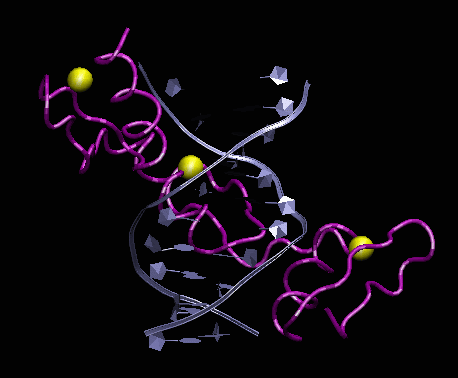
Biological systems chosen for this
study.
Left: crystal structure of Human four-helical domain of FRAP binding to
the complex FKBP12-rapamycin (PDB ID 3FAP). Right: Crystal structure of
a 3 zinc fingers domain from Zif268 in complex with DNA (PDB ID 1AAY).
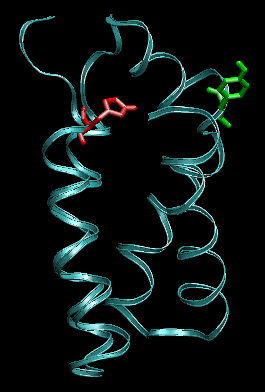
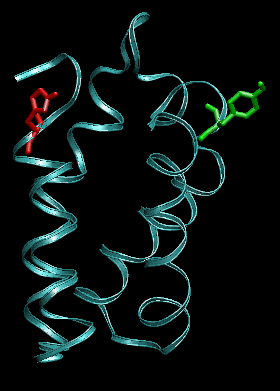
Snapshots before (left) and after 1ns (right) pulling between the residues indicated in green and red. The protein conformation is stable.
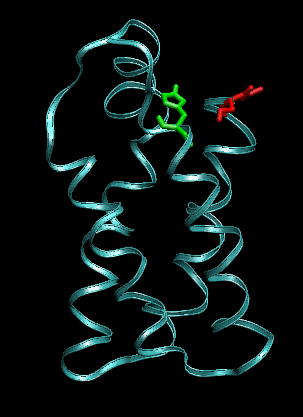

Snapshots before (left) and after 1ns (right) pulling between the residues indicated in green and red (other pair than shown previously). The protein starts to unfold.
References
[1] M.E. Chicurel, C.S. Chen and D.E. Ingber. Cellular control lies in the balance of forces. Curr Opin Cell Biol, 10:232-239,1998.
[2] Y. Sawada and M.P. Sheetz. Force transduction by Triton cytoskeletons. J Cell Biol, 156:609-615, 2002.
[3] M.J. Lang, P.M. Fordyce and S.M. Block. Combined optical trapping and single-molecule fluorescence. J Biol, 2:6, 2003.
The Stata Center, Building 32 - 32 Vassar Street - Cambridge, MA 02139 - USA tel:+1-617-253-0073 - publications@csail.mit.edu (Note: On July 1, 2003, the AI Lab and LCS merged to form CSAIL.) |