
Computational Design of Calcium-Sensitive Protein Sensors
David F. Green, Alan Jasanoff & Bruce Tidor
Summary
Computational protein design methods show great promise as a powerful tool for the development of novel reagents for the study of biological systems. We are furthering the development of these methods for application to the design of a calcium-sensitive probe for use in functional MRI studies of neural activity at the single-cell level. Taking the calmodulin–peptide binding system as an initial model, we design mutations to both binding partners so as to create a novel protein–peptide complex with minimal cross-reactivity to the wild-type complex. In this manner, we may develop a calcium sensing mechanism that, while based on calmodulin and sharing many properties with this evolved system, is orthogonal in its activity — the designed components will interact with one another, but not with the natural components. As a result, this reagent may be introduced into live organisms with minimal perturbations to the native cellular behavior.
Background
Recent advances in magnetic resonance imaging (MRI) methods have led to the possibility of using cellular-level contrast agents for "molecular functional MRI", with high resolution in both the spatial (50 μm ) and temporal (100 ms) domains [1]. A key requirement for these methods is the availability of molecular contrast agents that respond to functionally relevant changes in the intra-cellular environment. For the study of neural activity, cellular calcium ion concentrations provide a strong signal, and the Calmodulin (CaM) and CaM-binding peptide system provides a natural complex that associates and dissociates in a calcium sensitive manner. Re-engineering of the CaM complex to have an orthogonal interaction specificity relative to the natural system would improve the utility of this system as a probe for in vivo studies.
Over the past fifteen years, advances in computational methods for discrete conformational search as applied to the side-chain packing problem of protein design have led to remarkable successes in targeted design of isolated proteins [2]. However, the design of protein binding interfaces poses additional problems. One of the most significant of these is achieving specific binding. In order to successfully design a novel binding complex for use in vivo, the designed components must selectively bind to one another, while not interacting with an other components of the biological system. The question of how to efficiently and successfully design specifically-interacting protein complexes is directly addressed by this project. In addition, the direct relevance of the application to the advancement of biological knowledge is clear.
|
||
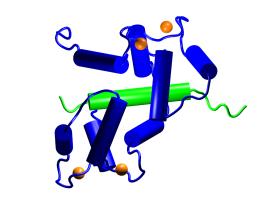
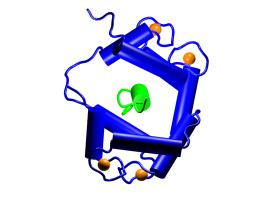
Overview of the CaM–M13
protein–peptide complex. The two viewpoints shown are
related by a 90° rotation about the vertical axis. CaM is shown
in blue, M13 in green, and Ca2+ ions in orange
[3]. |
Method of Approach
Several algorithms for highly efficient conformational search have been applied in the context of protein design, including the dead-end elimination (DEE) algorithm [4], and the A* tree search [5]. The DEE algorithm in particular has been shown to be incredibly useful in reducing the space of the conformational search problem, however it is plagued by one key limitation — it requires the use of a pair-wise selection (energy) function. Nonetheless, the power of the DEE algorithm, coupled with the immense complexity of the search space, makes the use of an approximate pair-wise energy function for the initial selection of structures essential. As measures of specificity are generally non-pairwise, as are important energetic components such as solvation, we focus on a hierarchical approach. Initial selection of likely low-energy states are selected using an approximate pair-wise function which allows us to harness the power of the DEE and A* algorithms. These states are then passed through a series of screening functions of increasing accuracy, and expense, passing on a reduced set of high-ranking candidates at each stage. In this way, the immense space of sequence and structural variability can be addressed efficiently, while maintaining two key properties — a high degree of accuracy in the energetic model used, and a high confidence of finding all solutions within a given cutoff of the global minimum.
Current Progress:
Our computational design protocol has been applied to several sites on the CaM–M13 binding interface, chosen through a visual analysis for mutable intermolecular interactions. These include regions where the desired specificity may be introduced through steric complementarity as well as those where charge-reversal modifications might be made. Initial calculations have suggested mutations operating through both of these mechanisms. As an example of steric reversal, the simultaneous replacement of M13 Trp4 with Phe (reducing the size of the peptide side chain) and CaM Phe92 with Trp (reducing the size of the protein binding pocket) suggests that this will be effective in reducing the affinity of non-desired complexes, while maintaining a near-native affinity for the mutant complex. Charge-reversal mutations are suggested involving M13 Lys19, where mutation to a Glu (replacing a positive charge with a negative charge) may be complemented by mutations to positive groups at complementary sites on CaM. A small set of these mutations have been experimentally tested, with promising results for the utility of this approach.
|
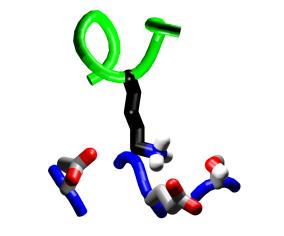
||||
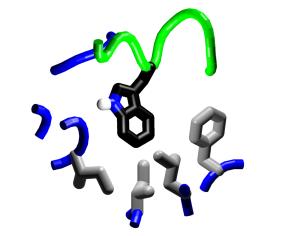
Steric complementarity.
The binding pocket of M13 Trp4 offers opportunities for specificity
through steric complementarity. |
||||
|
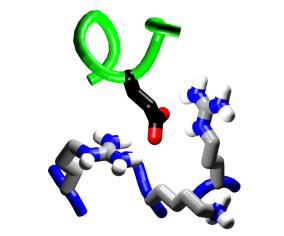
||||
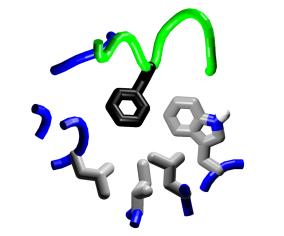
Charge reversal. Position
19 of M13, a lysine, provides an interesting location for consideration
of charge-reversal mutations. |
Future Directions
We are currently expanding the scope of our design methodology in several ways. These include explicit consideration of variant structures for non-desired complexes (decoys), and incorporation of the selectivity of binding criterion earlier into the search procedure. In addition, we are working through iterative cycles of calculations and experimental testing, so that we may use this data to refine our procedure.
References
[1] A. Jasanoff. Functional MRI using molecular imaging agents. Trends Neurosci. 28, pp. 120–126, 2005.
[2] B. I. Dahiyat and S. L. Mayo. De novo protein design: Fully automated sequence selection. Science 278, pp. 82–87, 1997.
[3] M.Ikura, G. M. Clore, A. M. Gronenborn, G. Zhu, C. B. Klee and A. Bax. Solution structure of a calmodulin–target peptide complex by multidimensional NMR. Science 256, pp. 632–638, 1992.
[4] J. Desmet, M. De Maeyer, B. Hazes and I. Lasters. The dead-end elimination theorem and its use in protein side-chain positioning. Nature 356, pp. 539–542, 1992.
[5] A. R. Leach and A. P. Lemon. Exploring the conformational space of protein side chains using dead-end elimination and the A* algorithm. Proteins 33, pp. 227–239, 1998.
The Stata Center, Building 32 - 32 Vassar Street - Cambridge, MA 02139 - USA tel:+1-617-253-0073 - publications@csail.mit.edu (Note: On July 1, 2003, the AI Lab and LCS merged to form CSAIL.) |