
Inverse Design of Molecular Libraries Enriched in Minimal Inhibitors of HIV-1 Protease
Michael D. Altman & Bruce Tidor
Introduction
Drug resistance continues to be one of the limiting factors in the treatment of HIV infection with protease inhibitors. In resistant HIV, the protease enzyme has mutated such that drug binding is reduced while enzymatic activity remains largely unaffected. In order to computationally design inhibitors that do not elicit resistance mutations, we have adopted a strategy called the minimal substrate shape hypothesis. It has been shown in several crystal structures of inactivated HIV-1 protease that substrates occupy a common minimal shape within the active site [1]. By targeting this minimal substrate shape with computational drug design techniques, we hope to create inhibitor molecules that will not result in the evolution of a resistant viral population because any mutation that disrupts inhibitor binding should also disrupt substrate binding and catalysis, making the virus non-viable.
To target the minimal substrate shape, we have focussed our efforts on designing libraries of molecules based on a known protease inhibitor molecular scaffold (Figure 1). This scaffold is the core of the clinically approved protease inhibitor amprenavir (Figure 2), which although potent initially, leads to the accumulation of resistance mutations and weakened binding. The goal of computational design on the amprenavir scaffold is to replace the functional groups (denoted by R) with alternative groups that cause the molecule to fit better within the minimal substrate shape while still making strong binding interactions. These molecules can then be synthesized and tested for binding against the wild-type protease as well as highly resistant protease mutants. If the compounds bind well to both, it will be suggestive of the effectiveness of the minimal substrate shape hypothesis. A further, more definitive test, is to apply the molecule to virus propagating in laboratory cell culture, and to examine the population for evolved resistance. Collaborations are in place to synthesize and study designed molecules in each of these modes.
Methods
The computational drug design method used to select functional groups for the scaffold is called inverse drug design. The inverse drug design procedure begins with a target shape, computes electrostatic and shape potentials inside this volume, and attempts to grow molecules that reproduce these potentials by means of combinatorial search. This technique differs from traditional drug design methods, such as docking [2], which take libraries of complete molecules or molecular fragments and try to fit them in the target site directly.
As input to the inverse drug design algorithm, the minimal substrate shape, generated from the crystal structures of inactive protease-substrate complexes, serves as the target. Fitting inside this shape is a hard constraint in the computational drug design process. Electrostatic and shape potentials are computed on a grid inside this volume, allowing for fast grid-based energy calculations. The amprenavir scaffold (Figure 1) is placed throughout the target shape in a discrete and flexible manner, and discrete libraries of functional groups are grown form the variable positions, denoted by R. The self and pairwise contributions to the binding energy are computed for each functional group and pair of functional groups through use of the grid potentials. Since the form of the energy function is pairwise, the search through functional group space for the best binding molecules is amenable to fast combinatorial search algorithms such as dead-end elimination (DEE) [3] and A* [4].
The energy function used during the combinatorial search phase is approximate, due to the pairwise approximation as well as computing all the grid potentials given a fixed target shape. To correct for these approximations, we use hierarchical energy functions, where the best molecules from the combinatorial search are progressively reevaluated in more accurate energy functions. After the hierarchical rescoring, we are left with a list of the highest ranked molecules in our best energy function, from which molecular libraries can be derived for synthesis and testing.
Progress
Inverse design has been successfully applied to the selection of functional groups for the amprenavir scaffold, and small libraries of 20-40 compounds have been developed. These libraries are currently being synthesized by our collaborators.
Future
In the future, the proposed small library of compounds will be synthesized and tested for binding in wild-type HIV protease as well as a few multi-drug resistant proteases in order to evaluate the minimal substrate shape hypothesis. If any compound shows broad binding to all mutational classes, it will be further subjected to tests such as in vitro passaging experiments to see if it is possible to directly evolve resistance against the compound. In addition, if many of the designed molecules are binders, it will serve to validate the inverse design method itself. Designed compounds that do not demonstrate binding are also important because they can reveal weaknesses in inverse design that can be improved.
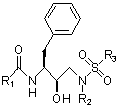
Figure 1: Amprenavir scaffold |
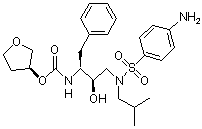
Figure 2: Amprenavir |
References
[1] M. Prabu-Jeyabalan, E. Nalivaika and C. A. Schiffer. Substrate shape determines specificity of recognition for HIV-1 protease: Analysis of crystal structures of six substrate complexes. Structure, 10:369-381, 2002.
[2] R. L. DesJarlais, R. P. Sheridan, J. S. Dixon, I. D. Kuntz and R. Venkataraghavan. Docking flexible ligands to macromolecular receptors by molecular shape. Journal of Medicinal Chemistry, 29:2149-2153, 1986.
[3] J. Desmet, M. Demaeyer, B. Hazes and I. Lasters. The dead-end elimination theorem and its use in protein side-chain positioning. Nature, 356:539-542, 1992.
[4] A. R. Leach and A. P. Lemon. Exploring the conformational space of protein side chains using dead-end elimination and the A* algorithm. Proteins: Structure Function and Genetics, 33:227-239, 1998.
The Stata Center, Building 32 - 32 Vassar Street - Cambridge, MA 02139 - USA tel:+1-617-253-0073 - publications@csail.mit.edu (Note: On July 1, 2003, the AI Lab and LCS merged to form CSAIL.) |