
Broad and Narrow Specificity in Molecular Design
Mala L. Radhakrishnan & Bruce Tidor
Introduction
Why are certain biological molecules naturally promiscuous in their binding interactions, able to bind with broad specificity to multiple targets, while other molecules are very selective, binding to only one partner with narrow specificity? What molecular interactions and chemical mechanisms dictate relative binding specificity? Finally, how can we actively incorporate binding specificity, whether broad or narrow, into the design of therapeutics? To address these questions we are studying how model and natural molecules use a variety of chemical mechanisms, including electrostatic interactions, molecular shape, and changes in titration state, to tailor their interactions with other molecules. In particular, we are analyzing determinants of binding specificity through a combination of exploring fundamental principles in simplified model systems together with an investigation of natural biological systems in which specificity plays a key role. These analyses will not only give us insight into characteristics that make molecules suited for broad or narrow specificity, but they will also allow us to further develop and apply useful molecular design heuristics and algorithms
Motivation
Understanding the molecular basis for binding specificity and developing methods to design molecules with tailored specificity will be of great use to the pharmaceutical community. Currently, the industry is faced with the challenge of designing safe and effective drug molecules that will bind to their targets with high affinity. However, considering only the primary binding interaction in the design process is not enough; one must also tune the binding specificity for the application at hand. In certain instances, there may exist molecules other than the target to which the drug molecule can bind, leading to undesirable side-effects. In these cases, the design process must steer toward the target and against such unwanted interactions. In other cases, the target might mutate rapidly and the designed drug should be able to bind to a broad range of related targets [1].
Our Approach
First, to address the question of how a molecule's structural characteristics can affect the specificity with which it binds to a spectrum of possible binding partners, we have developed a set of model compounds that retain much of the physical richness of real protein systems, but allow us to systematically vary such structural characteristics and study how such variations affect binding properties. Moreover, computations are particularly rapid for this system, which permits quick prototyping and statistical analysis of data. By systematically varying the charge distributions, shapes, sizes, and conformational flexibilities of such model molecules, we have gained valuable insight into how these properties can lead to either broad or narrow specificity. Moreover, the correlations in the data are in agreement with predictions of binding specificity from analytical electrostatic optimization theory in a continuum solvent [2][3]. With a firmer understanding of how structural characteristics of a molecule may make it a more or less specific binder in a biological environment, we are ready to begin synthesizing such insight into our current methods to design molecules with tailored specificity.
In addition to gaining an understanding of the molecular determinants of binding specificity, we have begun designing molecules with tailored specificity -- both narrow and broad:
Narrow Specificity:
The Erythropoietin system: Erythropoietin (Epo) is a hormone involved in the maturation of red blood cell precursors. Because Epo is produced by the kidney, patients with renal failure often develop anemia and rely on external sources of Epo, such as rHuEPO, for proper red blood cell development [4]. A current priority is to increase the half-life of these drugs so patients will not have to perform painful injections as often.
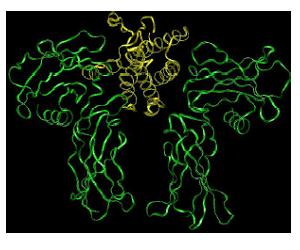
Figure 1. Cartoon representation of the Erythropoietin hormone bound to the extracellular portion of its receptor [5].
The pH-dependent binding specificity of a ligand to its receptor is often correlated with its in vivo half-life. We have identified residues on the Epo receptor (EpoR) that, based on computational predictions, confer narrow specific binding to Epo over a small pH range. Certain predicted mutations at these sites may serve as useful tools to help us understand how pH-dependent binding and the drug's cellular trafficking properties are related. In addition, using the same general technique as Sarkar et al. [6], we have identified potential mutations on Epo itself that might further narrow the binding specificity over a given pH range; such large pH-dependence in the binding might have positive implications on the drug's in vivo half life.
Broad Specificity:
HIV-1 protease and its inhibitors: HIV-1 protease cleaves protein chains to produce the active proteins necessary for viral activity. Its significance makes it an especially popular target for drugs to treat HIV patients. While there are many drugs that currently target HIV protease by binding in its active site, their effectiveness inevitably decreases as the protease mutates to become drug resistant. It is therefore crucial to account for existing and potential mutants of HIV protease in the design of an effective drug, while also designing against side reactions.
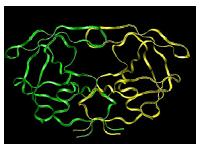
Figure 2. Cartoon representation of HIV protease [7].
We plan to directly target an ensemble of escape mutants, thus designing small molecules with "broad specificity." Because of the large number of potent escape mutants, it may not be possible to find one molecule that "covers" (i.e. binds well to) all the escape mutants. Therefore, we have developed a simple method for optimally "tiling the space of escape mutants" -- finding the minimal set of drug molecules that ensures that every escape mutant gets bound well. Effectively, combining this functionality with our current drug design capabilities will allow us to design "optimal drug cocktails" for rapidly mutating targets.
Research Support
This work is partially being funded by the National Institutes of Health and by a Department of Energy Computational Science Graduate Fellowship (DE-FG02-97ER25308)
References:
[1] E. Friere. Designing drugs against heterogeneous targets. Nature Biotech, 20:15-16, 2002.
[2] E. Kangas and B. Tidor. Electrostatic specificity in molecular ligand design. J. Chem Phys, 112:9120-9231, 2000.
[3] L.-P. Lee and B. Tidor. Optimization of electrostatic binding free energy. J. Chem. Phys., 106:8681-8690, 1997.
[4] S. Elliott et al. Enhancement of therapeutic protein in vivo activities through glycoengineering. Nature Biotech., 10:1-8, 2003.
[5] R. S. Syed et al. Efficiency of signalling through cytokine receptors depends critically on receptor orientation. Nature, 395:511-516, 1998.
[6] C. Sarkar, K. Lowenhaupt, T. Horan, T. C. Boone, B. Tidor, D. A. Lauffenburger. Rational cytokine design for increase lifetime and enhanced potency using pH-activated "histidine switching". Nature Biotech., 10:1-6, 2002.
[7] J.L. Martin et al. Molecular Recognition of Macrocyclic Peptidomimetic Inhibitors by HIV-1 Protease Biochemistry, 38:7978-7988, 1999.
The Stata Center, Building 32 - 32 Vassar Street - Cambridge, MA 02139 - USA tel:+1-617-253-0073 - publications@csail.mit.edu (Note: On July 1, 2003, the AI Lab and LCS merged to form CSAIL.) |