| Technical Reports | Work Products | Research Abstracts | Historical Collections |
![]()
|
Research
Abstracts - 2006
|
|
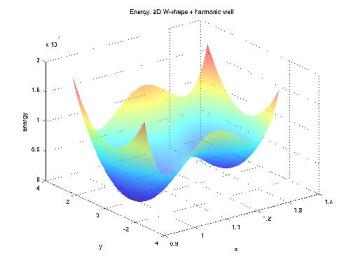
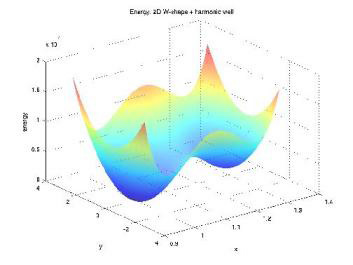
Molecular Transduction of Mechanical Signals: Simulation Study of Force-induced Conformational ChangeAurore Zyto, Roger D. Kamm & Bruce TidorMechanical signals have been shown to regulate various biological behaviors, including growth, differentiation, apoptosis and cell motility [1]. The details of the process by which these cues result in cellular change are largely unknown. One hypothesis is that key protein molecules act as mechanosensors and mechanotransducers; the combined effect of these functions is to convert a mechanical signal into a chemical one that connects to cellular biochemistry and downstream effectors. One mechanism for achieving mechanotransduction is through force-driven conformational change, and a more generalized mechanism is force-modulated binding affinity [2]. In this work, we apply computational modeling to investigate key features of molecular events involved in mechanotransduction. The computational studies correspond to biological systems that can be probed with single-molecule experiments. Our focus is on elucidating biomolecule structure-function relationships and the processes by which they change in response to mechanical force. Progress in development of general approaches to study force-induced changes in the structural properties and associated biochemical activity of proteins is illustrated. MethodsTo assess the force effect on protein biophysics, we wish to compute the change in binding free energy of a given protein-ligand complex as we apply a force to one of its components. A first step towards this goal is to estimate the free energy change of a system under force application. We adapted Hamiltonian switching methods to the special case of a force as a perturbing factor [3]. Overall accuracy and performance of the method are tested with simple systems, for which the exact free energy change can be computed. We address specific questions that are relevant for concrete application to protein systems, such as the impact of the number of dimensions or local minima on the convergence properties (Figure 1). We identified selected biological systems of interest. Four-helix bundles are a simple, common motif of protein structure of regular topology. Several focal adhesion proteins, including some potential mechanosensors, are made up of those domains. We're currently focusing our efforts on a member of this cellular component: FAT domain of Focal Adhesion Kinase binding to a Paxillin peptide (Figure 2). We model the protein in its bound and unbound states from the available crystal structure. Molecular Dynamics simulations under force application enable us to address two main questions. First, we examine the structural effects by applying a constant force; this is a useful step to analyze the differences in behavior for the protein in its bound and unbound states. Second, free energy simulations are performed with a time-varying force, leading to estimates of the binding free energy as a function of the applied force. ProgressPerformance of our free energy method was successfully tested on harmonic systems with up to 100,000 dimensions. The overall behavior as a function of the number of dimensions looks promising. Simulations on energy landscapes with many local minima help us gain insight into the computational difficulties that arose for the protein system. General conditions to ensure confidence in the estimates are being developed. FutureMethodological advances will facilitate efficient application to protein systems. The nature, extent, and robustness of the force-response will be assessed through study of various pulling directions. Differences in behaviors of the bound and unbound states could be confirmed with single-molecule experiments and may shed light into a much unknown biological phenomenon.
FundingThis work is supported by the National Institutes of Health. References:[1] M.E. Chicurel, C.S. Chen and D.E. Ingber. Cellular control lies in the balance of forces. Curr Opin Cell Biol, 10:232-239, 1998. [2] Y. Sawada and M.P. Sheetz. Force transduction by Triton cytoskeletons. J Cell Biol, 156:609-615, 2002. [3] M. Watanabe and W.P. Reinhardt. Direct dynamical calculations of entropy and free energy by adiabatic switching. Phys Rev Lett, 65:3301-3304, 1990. |
||||||
|