| Technical Reports | Work Products | Research Abstracts | Historical Collections |
![]()
|
Research
Abstracts - 2006
|
|
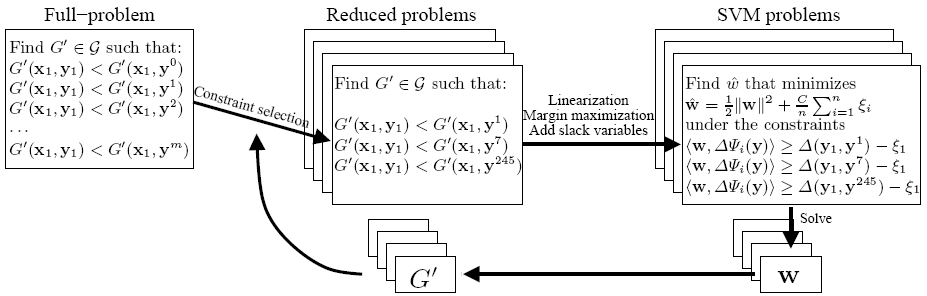
Learning Biophysically-Motivated Parameters for Alpha Helix PredicationBlaise Gassend, Charles W. O'Donnell, William Thies, Andrew Lee, Marten van Dijk & Srinivas DevadasIntroductionOur goal is to develop a state-of-the-art protein secondary structure predictor, with an intuitive and biophysically-motivated energy model. The lack of experimentally determined free energy values makes it difficult to design accurate cost functions that can be optimized by predictors. Our technique uses a cost function comprised of unknown parameters, and applies Support Vector Machines (SVMs) to learn parameters that correctly predict known protein structures. So far, we have focused on the prediction of all-alpha proteins and have shown that a model with 302 parameters can achieve a Qalpha value (percent of correctly predicted residues) of 77.6% and a SOV99alpha (see [1]) value of 73.4%. As detailed in a recent technical report [2] and poster [3], these performance numbers are among the best for techniques that do not rely on additional databases (for example, multiple sequence alignments). MethodOur method assumes that a protein's secondary structure can be found by minimizing a free-energy function G' that is computed as a sum of elementary free-energies. For example, an elementary free-energy might represent the energetic cost of a given residue appearing in an alpha helix. The predicted structure minimizes the sum of these costs. For fixed values of the free-energy parameters, there are well-known algorithms to perform such a minimization (for example, dynamic programming). Thus, our main task is to find the unknown elementary free-energies by using a database of known protein structures. A general Support Vector Machine algorithm has been proposed that can be applied to this [4]. The key ideas of the algorithm are illustrated in the figure below. First, the problem is converted into an exponentially large system of inequalities that the elementary free-energies must satisfy: for each sequence xi, the correct secondary structure yi must have a lower free-energy G'(xi, yi) than for any of the incorrect secondary structures yj. Next, a tractable subset of these inequalities is selected. This subset may not have any solutions, because there might not exist a set of free-energies that is compatible with the whole database of training structures. Alternately, if the problem does have solutions, it will probably have many. The SVM techniques of margin maximization and slack variables are used to translate the reduced problem into a quadratic program that has a unique solution. The quadratic program is solved to produce a candidate set of elementary free-energies, which is used by a structure predictor (not shown) to predict a new structure for each sequence xi. If any of these structures are incorrect, they are added to the subset of incorrect structures yj, a new quadratic program is generated, and a new candidate solution is found. We repeat these steps until all generated structures are correct (or in practice, a suitable termination condition is satisfied).
ResultsWe have applied this method to the case of all-alpha protein secondary structure prediction. We worked with a set of 300 non-homologous all-alpha proteins taken from EVA's largest sequence-unique subset of the PDB at the end of July 2005. For each run of our algorithm, we randomly selected a 150 protein training set and an independent 150 protein test set. The training set is used to learn the elementary free-energies, and the test set is used to evaluate the result. Our predictor minimizes the free-energy function G' using the Viterbi algorithm on a simple 7-state Finite State Machine. The following table summarizes our results. The prediction accuracy is competitive with other state-of-the-art predictors that do not rely on sequence alignment data. Moreover, while some techniques require upwards of 10,000 parameters, our predictor uses only 302 parameters in the form of elementary free-energies [2].
ConclusionsThis work is a promising first pass at using SVM techniques to find the elementary free-energies needed to predict protein secondary structure. The method we use is general and can be extended beyond the all-alpha case described here. In future work, we plan to extend this method to super-secondary structure prediction, generating contact maps of individual hydrogen bonds in beta sheets. References:[1] A. Zemla, Ceslovas Venclovas, K. Fidelis, and B. Rost. A Modified Definition of Sov, a Segment-Based Measure for Protein Secondary Structure Prediction Assessment. Proteins, 34(2), 1999. [2] Blaise Gassend, Charles W. O'Donnell, William Thies, Andrew Lee, Marten van Dijk, Srinivas Devadas. Secondary Structure Prediction of All-Helical Proteins Using Hidden Markov Support Vector Machines MIT CSAIL Technical Report 1003 (MIT-LCS-TR-1003) [3] Blaise Gassend, Charles W. O'Donnell, William Thies, Andrew Lee, Marten van Dijk, Srinivas Devadas. Learning Biophysically-Motivated Parameters for Alpha Helix Prediction. Poster at the 10th Annual International Conference on Research in Computational Molecular Biology, Venice Lido, Italy, 2006. [4] I. Tsochantaridis, T. Hofmann, T. Joachims and Y. Altun. Support Vector Machine Learning for Interdependent and Structured Output Spaces. In Proceedings of the 21st International Conference on Machine Learning, Banff, Canada, 2004. |
||||||||||||||||||||||||||||
|