
| Technical Reports | Work Products | Research Abstracts | Historical Collections |
![]()
|
Research
Abstracts - 2006
|
|
Combinatorial Computational Design of Cytosine PhosphoribosyltransferaseSurasak Chunsrivirot, Michael D. Altman, James R. Williamson, & Bruce TidorWhatTo produce an enzyme with a novel function through directed evolution, a library is created mostly by randomization of the DNA codons of an enzyme sequence. However, the size of the library might be extremely large and hence, difficult or impossible to screen and select, especially if the desired active site of the enzyme is large and complex. To reduce the size and improve the quality of libraries, we used rational design to develop an approach to produce sets of computed sequences to be enriched for a novel enzymatic function, cytosine phosphoribosyltranferase (CPRTase) activity. CPRTase catalyzes the formation of cytidine 5'-monophosphate (CMP) from cytosine and phosphoriboyslpyrophosphate (PRPP). The formation of CMP from PRPP and cytosine is shown in Figure 1. To date, no natural CPRTase has been purified to confirm its existence, and no structural or genomic data of this natural enzyme has been deposited in the National Center for Biotechnology Information (NCBI) database. If one can produce CPRTases, they may be particularly useful for the incorporation of spin labels into polynucleotides for biophysical study. 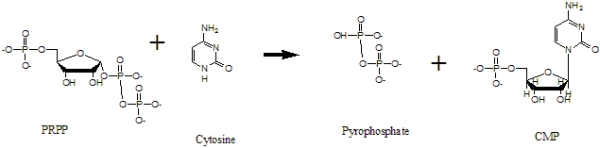 Figure 1. The formation of CMP from PRPP and cytosine HowStructure SelectionTo create Cytosine Phosphoribosyl Transferase, we need promising crystal structures of Pyrimidine/Purine Phosphoribosyltransferases to use as base structures in our design approach. We used the following criteria in structure selection: a structure should contain the substrates (base ring, PRPP) and cofactors (metal ions); it should contain side-chain interactions (H-bonds) with the base; the base should have functional groups that are similar to cytosine; the active site should be enclosed by a loop. After exploring structures deposited in the Protein Data Bank, we selected two structures: 1TC2 (hypoxanthine phosphoribosyltransferase) and 1L1R (adenine phosphoribosyltransferase). 1TC2 is a hypoxanthine phosphoribosyltransferase that catalyzes the formation of inosine 5'-monophosphate (IMP) from hypoxanthine and phosphoribosylpyrophosphate (PRPP); it is an asymmetric dimer from the protozoan parasite Trypanosoma cruzi. This structure contains PRPP, an analogue of the substrate (7-hydroxy[4,3-d]pyrazolopyrimidine) (HPP), Mg & Mn ions. It has one open and one closed subunit; the active site is located between the core and hood domains of the enzyme. This enzyme has a flexible loop closeing the active site upon binding with substrates. The structure of 1TC2 is shown in Figure 2.
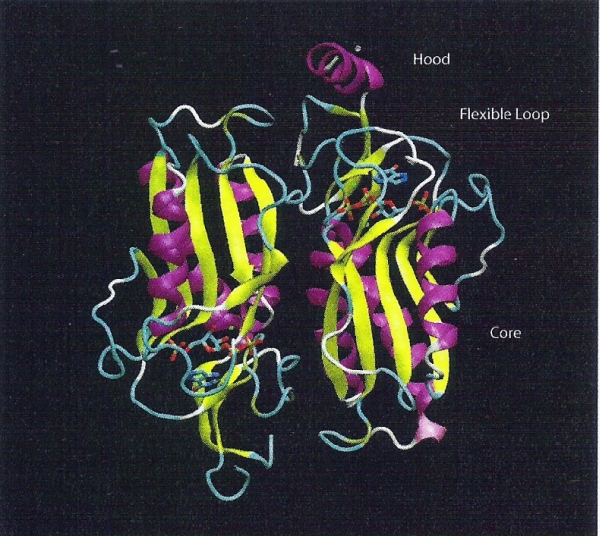 Figure 2. The structure of 1TC2 (HPRTase) is represented as cartoon and colored by their structures. 7HP, PRPP, Mg and Mn are represented as licorice and colored by their element type. The open subunit is shown on the left and the closed subunit is shown on the right. The hood and the core domains surround the active site. 1L1R is an Adenine Phosphoribosyltransferase that catalyzes the formation of AMP from Adenine and PRPP. It is a symmetric homodimer.The catalytic residue is on the loop over the active site pocket, and the loop will be closed when the substrate binds to the active site. This enzyme was co-crystalized with 9-deazaadenine (9DA), Mg ion, PRPP, and its sturcture is shown in Figure 3.
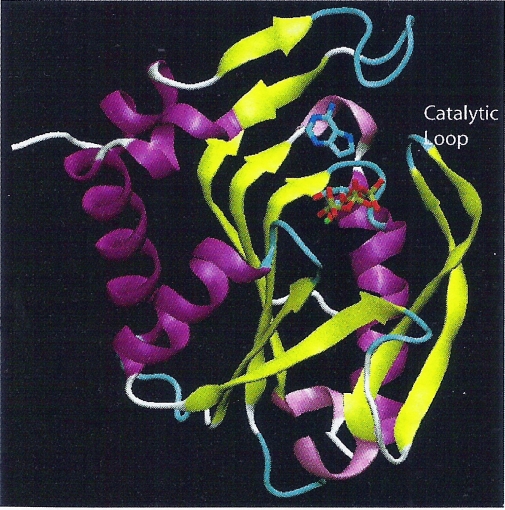 Figure 3. The structure of 1L1R
(APRTase) is represented as cartoon and colored by their structures.
9DA, PRPP and Mg2+ are represented as licorice and colored by their
element type. The catalytic loop fully closes the active site.
Design ApproachBecause enzymes, naturally, are good at binding a transition state of a substrate so that they can catalyze the formation of the product effectively, we proposed the resonance structures of cytosine that are likely to be the transition states by proposing a possible mechanism (resulting in one tautomer of cytosine). We built this resonance form into the active sites of 1TC2 and 1L1R in the same position and orientation as those of existing base, and we used these structures as inputs of the design. Moreover, we selected the designed positions and possible amino acids at a specific position based on the knowledge from literature and from molecular modeling. Since there are more than thousands of possible sequences resulting from the output of the software, we used the following criteria in selecting promising sequences. The output structures should have a new catalytic residue that is close enough to abstract a proton from the appropriate cytosine tautomer, a residue that can make hydrogen bonds to -NH2 of cytosine, an aromatic residue that is in the same plane and close to the base ring of cytosine and residues that are able to fill in spaces that the six-membered ring part of the purine base used to reside. Protein libraries are then derived from these promising sequences. Finally, DNA libraries are derived from these protein libraries. ProgressWe have completed two designs on 1TC2 and 1L1R (each with one tautomer of cytosine), selected promising structures/sequences, derived the protein and DNA libraries from these designed sequences. Now, we are performing additional analysis on the stability of designed CPRTase’s active site, using stochastic boundary molecular simulation and the particle mesh Ewald method. Research SupportThis research was supported by MIT's Undergraduate Reseach Opportunities Program, the John Reed Fund, and the Paul E. Gray Fund. References[1] P. J Focia, S. P. Craig III and A. E. Eakin: Approaching the Transition State in the Crystal Structure of a Phosphoribosyltransferase. In Biochemistry, 37, pp. 17120,1998 [2]W. Shi, Anne Sarver, Ching Wang, Kelly Tanaka, Kelly and Steven Almo. Closed Site Complexes of Adenine Phosphoribosyltransferase from Giardia Lamblia Reveal a Mechanism of Ribosyl Migration. In J.Biol.Chem., 277 pp. 39981, 2002 [3]S. P. Craig III and A. E. Eakin. Purine Phosphoribosyltransferases. In The Journal of Biological Chemistry 275, pp. 20231-20234, 2000 [4]Kadziola, Anders., Neuhard, Jan & Larsen, Sine. Structure of product-bound Bacillus caldolyticus uracil phosphoribosyltransferase confirms ordered sequential substrate binding. In Biological Crystallography, D58, pp. 936-945, 2002. |
|||
|