| Technical Reports | Work Products | Research Abstracts | Historical Collections |
![]()
|
Research
Abstracts - 2006
|
|
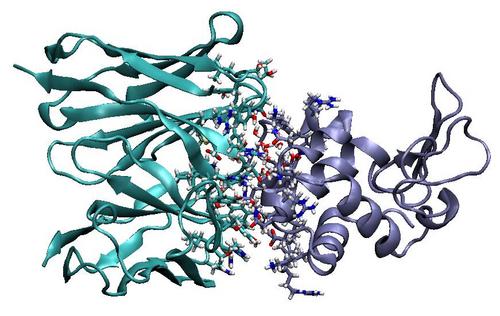
Antibody Affinity Maturation Using Computational Protein DesignShaun M. Lippow, K. Dane Wittrup & Bruce TidorIntroductionThe biochemical process of two proteins in solution forming a single bound complex is a basic molecular event in biology with relevance to the study of biological systems and the development of treatments for disease. The strength, or affinity, of a binding reaction is of critical importance for drug design. The aim of this project is to develop and apply computational protein design tools for engineering high-affinity protein interactions. The problem of main interest is to predict side chain mutations that will improve the binding affinity in an existing, sub-optimal protein complex. MotivationAntibodies and antibody fragments are an important class of proteins used as drugs and as reagents in biological diagnostics. Therapeutic antibodies can function as binding antagonists or as vehicles for specific drug delivery. Engineering an antibody for high-affinity binding to a protein of interest is often a main development step. Computational protein design and directed evolution methods provide complementary and potentially synergistic approaches to protein engineering. Computations can in principle search a vastly larger sequence space, albeit with an approximate energy description serving as a surrogate for biochemical function. Directed evolution is limited by experimental library size, with random mutagenesis generally covering most single mutations and only sampling larger combinations of mutations. Evolution methods are proficient at accumulating a series of additive mutations, but are less adept at identifying multiple, cooperative mutations. One of our interests is in exploring how much more would be gained by covering all double or triple mutations, or in other words, how often a pair or more of mutations improves binding affinity where each mutation alone is neutral or detrimental to binding. MethodsOur approach to protein design begins with an atomic-level model from x-ray crystallography of the initial antibody in complex with the protein of interest. For a given protein sequence and conformation, the free energy can be calculated using a physics-based energy function. The objective is to minimize the change in free energy on binding with respect to both side chain identity and conformation. The conformational space is generated using a library of discrete side chain rotamers from a statistical database and a fixed protein backbone. The problem is complicated by the fact that in reality, protein conformation can change upon binding, and each protein will adopt the conformation that minimizes the free energy of its current state. In addition, the optimization is subject to the biologically relevant constraint that a designed protein be stably folded, independent of binding. Packing and electrostatic effects are co-optimized using a hierarchical search that successively applies more accurate, yet more computationally expensive energetic models. In the first level of the search hierarchy, an approximate, pairwise-additive energy function amenable to the dead-end elimination and A* algorithms is used to efficiently prune much of the space [1, 2, 3]. The global optimum and all near-optimal solutions within an energy cutoff are kept from this stage. The pairwise-additive energy function used is from the CHARMM force-field and includes van der Waals interactions, covalent bonding terms, and simplified electrostratics with a distance-dependent dielectic constant [4]. Successive refinement steps enumerate the remaining list of candidate structures using, for example, Poisson-Boltzmann continuum electrostatics [5] and a continuum van der Waals model [6]. ResultsWe are investigating the model antibody, D1.3, which binds its antigen, hen egg lysozyme (HEL), with low nanomolar affinity [7]. We developed and applied our computational tools to predict the effect of all possible single mutations among the 61 complementarity determining region sites in D1.3. We found that most single mutations are predicted to be neutral or unfavorable and only a few variants are predicted to yield marginal improvements. We have experimentally tested some of the predictions of small improvement and validated the computations. Figure 1. Cartoon representation of the crystal structure of D1.3 bound to HEL, with D1.3 in cyan and HEL in iceblue. Side chains near the binding interface are shown in detail. Future WorkWork is underway to examine whether substantial, non-additive affinity enhancements are available through larger sets of multiple mutations. In addition, different antibody systems are being investigated. Research SupportThis work is supported in part by an NSF graduate fellowship to SML and the National Insititutes of Health. References:[1] J. Desmet, M. De Maeyer, B. Hazes, and I. Lasters. The dead-end elimination theorem and its use in protein side-chain prediction. Nature, 356:539-542, 1992. [2] D. B. Gordon, G. K. Hom, S. L. Mayo, and N. A. Pierce. Exact rotamer optimization for protein design. J. Comp. Chem., 24:232-243, 2003. [3] A. R. Leach and A. P. Lemon. Exploring the conformational space of protein side chains using dead-end elimination and the A* algorithm. Proteins, 33:227-239, 1998. [4] B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comp. Chem., 4:187-217, 1983. [5] M. K. Gilson and B. Honig. Calculation of the total electrostatic energy of a macromolecular system: solvation energies, binding energies, and conformational analysis. Proteins, 4:7-18, 1988. [6] R. M. Levy, L. Y. Zhang, E. Gallicchio, and A. K. Felts. On the nonpolar hydration free energy of proteins: surface area and continuum solvent models for the solute-solvent interaction energy. J. Am. Chem. Soc., 125:9523-9530, 2003. [7] T. N. Bhat, G. A. Bentley, G. Boulot, M. I. Greene, D. Tello, W. Dall'Acqua, H. Souchon, F. P. Schwarz, R. A. Mariuzza, and R. J. Poljak. Bound water molecules and conformational stabilization help mediate an antigen-antibody association. Proc. Natl. Acad. Sci., 91:1089-1093, 1994. |
|||
|