
| Technical Reports | Work Products | Research Abstracts | Historical Collections |
![]()
|
Research
Abstracts - 2006
|
|
Inverse Design of Paired Compounds to Test the Substrate Envelope Hypothesis in HIV-1 Protease Drug ResistanceMichael D. Altman & Bruce TidorIntroductionDrug resistance continues to be one of the limiting factors in the treatment of HIV infection with protease inhibitors. In resistant HIV, the protease enzyme has mutated such that drug binding is reduced while enzymatic activity remains at viable levels. In order to computationally design inhibitors that do not elicit resistance mutations, we have adopted a strategy called the minimal substrate shape hypothesis. It has been shown in several crystal structures of inactivated HIV-1 protease that substrates occupy a common minimal shape within the active site [1]. By targeting this minimal substrate shape with computational drug design techniques, we hope to create inhibitor molecules that will not result in the evolution of a resistant viral population because any mutation that disrupts inhibitor binding should also disrupt substrate binding and catalysis, making the virus non-viable. In order to fully test the hypothesis, we also design pairs of compounds (Figure 1), with one that stays inside the envelope and one that exceeds the envelope by changing a small part of the molecule. If the compounds that exceed the envelope are prone to resistance, while the ones that remain inside are not, then the substrate envelope hypothesis can be confirmed as a design principle in both a positive and negative direction. ProgressSo far, a small library of 15 compounds designed to stay within the substrate envelope were designed and synthesized. These compounds were derived from a chemical scaffold similar to the clinical inhibitor amprenavir and a set of functional groups from commercial catalogs. Of these computationally designed compounds, four bind tighter than 50 nM to the wild-type protease. In addition, these compounds have been tested for binding against a panel of drug-resistant proteases in which they lose little affinity compared to wild type. This demonstrates that the substrate envelope hypothesis is effective in the positive direction, meaning that compounds designed to stay inside have good resistance profiles. A second-round library has been recently proposed that contains paired compounds that exceed the envelope as well as compounds that remain inside but will hopefully have higher affinity. This library is currently being synthesized and tested by our collaborators. MethodsThe computational drug design method used to develop inhibitors within and exceeding the substrate envelope is called inverse drug design. The inverse drug design procedure begins with a target shape, computes electrostatic and shape potentials inside this volume, and attempts to grow molecules that reproduce these potentials by means of combinatorial search. This technique differs from traditional drug design methods, such as docking [2], which take libraries of complete molecules or molecular fragments and try to fit them in the target site directly. As input to the inverse drug design algorithm, the minimal substrate shape, generated from the crystal structures of inactive protease-substrate complexes, or a maximal shape, filling the active site completely for paired compounds, serves as the target. Designed molecules must fit inside these shapes, and interact favorably with the binding potentials. In order to find molecules that achieve these goals optimally, we employ a pairwise binding energy model and fast combinatorial search algorithms such as dead-end elimination (DEE) [3] and A* [4]. The best molecules from the combinatorial search are then re-evaluated in more sophisticated energy models that are harder to search and take longer to compute. FutureIn the future, the second-round library will be synthesized and tested. Hopefully, this library will contain compounds that bind tighter to the wild-type protease while staying inside the substrate envelope, and have broad specificity across drug-resistant proteases. Paired compounds that differ slightly from these compounds but exceed the envelope will hopefully have poor resistance profiles in order to confirm the substrate envelope hypothesis in the negative direction. The best in-envelope compounds from this second-round library will also be subjected to in vitro passaging experiments to see if it is possible to directly evolve resistance against them. 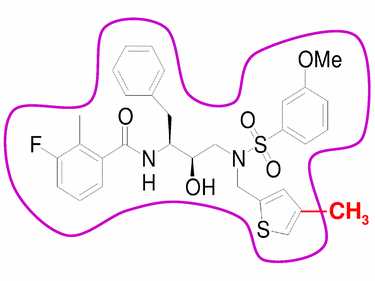
Figure 1: Paired compound example. The base compound (black) fits inside the substrate envelope (purple). When a methyl group (red) is added to the compound, it exceeds the envelope. Funding:This work is supported by the National Institutes of Health (NIH). References:[1] M. Prabu-Jeyabalan, E. Nalivaika and C. A. Schiffer. Substrate shape determines specificity of recognition for HIV-1 protease: Analysis of crystal structures of six substrate complexes. Structure, 10:369-381, 2002. [2] R. L. DesJarlais, R. P. Sheridan, J. S. Dixon, I. D. Kuntz and R. Venkataraghavan. Docking flexible ligands to macromolecular receptors by molecular shape. Journal of Medicinal Chemistry, 29:2149-2153, 1986. [3] J. Desmet, M. Demaeyer, B. Hazes and I. Lasters. The dead-end elimination theorem and its use in protein side-chain positioning. Nature, 356:539-542, 1992. [4] A. R. Leach and A. P. Lemon. Exploring the conformational space of protein side chains using dead-end elimination and the A* algorithm. Proteins: Structure Function and Genetics, 33:227-239, 1998. |
|||
|