
| Technical Reports | Work Products | Research Abstracts | Historical Collections |
![]()
|
Research
Abstracts - 2006
|
|
Comparative Gene Identification in Mammalian, Fly, and Fungal GenomesMichael F. Lin & Manolis KellisIntroductionA major challenge in analyzing any genome is the reliable identification of protein-coding genes. Complex genomes present a very low signal-to-noise ratio for gene identification; for example, only about 1.5% of the human genome sequence is protein coding. As a result, purely computational gene identification programs have achieved only limited success in complex genomes such as the human. The construction of gene catalogues for these species has instead relied primarily on expensive, error-prone mass sequencing of mRNA transcripts from cell cultures. A promising new approach for the identification of functional elements such as genes in the human and other genomes is the comparative analysis of the genomes of several related species. Because mutations in functional sequence are likely to be disfavored by natural selection, functional elements tend to be more strongly conserved across related genomes than nonfunctional sequence, which is subject to random mutation. We are developing computational methods for analyzing alignments of orthologous sequence from several related species in order to detect the distinctive signature of protein-coding sequence conservation and evolution. We are applying these methods to genomes from a wide variety of species, in order to both discover new genes and refine existing gene annotations. Ultimately, we hope to use our system to construct a complete set of conserved protein-coding genes for each of these species. Related workThe most successful approaches for purely computational gene identification in a single genome have relied on complex probabilistic models. These systems use statistical properties of protein-coding sequence and specific sequence signals such as splice sites in order to determine the most likely parse of a DNA sequence into protein-coding exons and noncoding regions. In the human genome, these systems have proven unreliable; for example, GENSCAN [1], the most widely used of these systems, makes predictions with a false positive rate of around 40%. Recently, these probabilistic models have been extended to additionally consider sequence conservation across a few related genomes, greatly improving their predictive accuracy [2-5]. However, these approaches are complex, requiring tens of thousands of parameters, and the types of signals they can consider are restricted by strong Markov assumptions in the model framework. It is unclear how well they will scale to use the larger numbers of genomes that are rapidly becoming available, or potentially informative sequence signals, such as splicing regulatory motifs, that are still being discovered. MethodsWe are developing an alternative approach to comparative gene identification based on direct evaluation and classification of candidate protein-coding intervals in sequence alignments. We use direct metrics of the distinctive evolutionary signatures of protein-coding sequence, including the strong pressure to preserve the reading frame of codon translation, the preference for synonymous codon substitutions, and substitutions preserving amino acid properties. We use these measurements to classify candidate sequence intervals as protein-coding or not, using standard classification algorithms such as support vector machines and random forests. In order to perform full de novo gene prediction, we simply enumerate all possible coding regions (exons) in the sequence alignment by identifying sequence signals such as start codons, stop codons, and splice sites, and apply our classifier to each interval. We find that our methods perform comparably to probabilistic approaches when measured using summary statistics on test sets such as the human ENCODE regions. Our system also enjoys several practical advantages over generative probabilistic models in terms of flexibility; for example, we can easily apply our classifier to existing gene annotations, and thus directly evaluate the conservation of known genes. We have also adapted our system to identify unusual events such as stop codon readthrough, a rare but essential phenomena that violates normal models of gene structure. 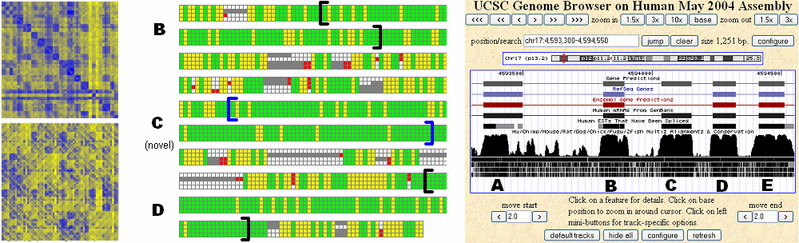
ResultsHuman: In the human genome, we have identified nearly 3,000 novel protein-coding exons for which there is strong comparative evidence, using alignments with the dog, mouse, and rat genomes. These are likely to correspond to previously unknown alternatively spliced exons as well as potentially hundreds of entirely novel genes. A sample of these was tested and confirmed using RT-PCR. Our analysis also casts doubt on hundreds of current gene annotations, including a small number in the well-annotated ENCODE regions. In addition to supporting or rejecting entire genes, our approach suggests many corrections to existing gene annotations, such as adding or removing individual exons. Fruit fly: In D. melanogaster, an important model organism, we have likewise used alignments with up to 8 other Drosophila species to identify more than 500 novel exons, both within existing gene annotations and in entirely novel regions of the genome. Our analysis also suggests that at least 500 presently annotated genes are not real; most of these are single-exon annotations which indeed have marginal support from experimental data. Our analysis is contributing to improvements in the official fly genome annotation through an ongoing collaboration with FlyBase. Fungi: We are using our methods to refine the gene annotations for C. albicans, a common human pathogen, and to assist in the initial annotation of several newly sequenced Candida species, including C. tropicalis, C. guilliermondii, and C. lusitaniae. In the yeast S. cerevisiae, we use alignments with other Saccharomyces species to analyze short genes (25-100 amino acids) that have previously been overlooked by comparative and other methods. References:[1] C. Burge and S. Karlin. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268:78-94, 1997. [2] I. Korf, P. Flicek, D. Duan, and M.R. Brent. Integrating genomic homology into gene structure prediction. Bioinformatics 17 Suppl 1:S140-148, 2001. [3] G. Parra, P. Agarwal, J.F. Abril, T. Wiehe, J.W. Fickett, and R. Guigo. Comparative gene prediction in human and mouse. Genome Res. 13:108-117, 2003. [4] A. Siepel and D. Haussler. Computational identification of evolutionarily conserved exons. Prec 8th 177-186, 2004. [5] S.S. Gross and M.R. Brent. Using multiple alignments to improve gene prediction. Prec 8th 375-388, 2005. |
||||
|