
| Technical Reports | Work Products | Research Abstracts | Historical Collections |
![]()
|
Research
Abstracts - 2006
|
|
Ortholog and Paralog Detection with Mulitple Complete GenomesMatt Rasmussen & Manolis KellisAbstractThe first challenge in comparative genomics is to reliably determine orthologous genes across multiple species. Current approaches work well with pairs of species but accumulate inaccuracies as more relatives are considered [1,2]. Hence, new methods are necessary to achieve accurate and reliable alignments of the upcoming mammalian, fly, and fungal species. We have developed and prototyped a new phylogenetic reconstruction method SINDIR, Species INformed DIstance-based Reconstruction. SINDIR finds the maximum likelihood gene tree using the known species and pair-wise distance estimates between gene sequences. The algorithm is designed specifically for the phylogenomic problem [3], namely the use of phylogeny to reconstruct the ancestry of all genes within several complete genomes. The resulting phylogeny can be used to make reliable ortholog assignments across several species, which is useful in comparative studies and also sheds light on the evolution of the species. ApproachWith a distance-based phylogenetic approach, it is possible to efficiently reconstruct phylogenies of thousands of gene families, some as large as hundreds of genes, in a reasonable amount of time. However, existing distance-based approaches, such as Neighbor Joining [4], often construct erroneous gene trees due to long branch attraction. Often these errors produce gene trees that disagree with the known species tree and thus erroneously infer gene duplications and losses. The key insight of our algorithm is to use the known species tree during the gene tree construction in order to identify unlikely inferred gene duplications and losses. The algorithm is given as input a rooted species tree topology and a small subset of genes known to be orthologous within the species of interest. Given these inputs, the algorithm computes a density estimation of the distribution of phylogenetic trees for true orthologs. Once the distribution parameters are learned, the algorithm takes a distance matrix for a family of genes with unknown orthology. In a search constrained by the distance matrix, the algorithm finds the maximum likelihood tree according to the learned distribution. Using reconciliation, the orthology of the gene family can be inferred. ResultsUsing highly confident orthologs from syntenic alignments of dog, human, mouse, and rat, we were able to evaluate the performance of SINDIR (Figure 1). On a dataset of 1800 gene families with topologies both agreeing and disagreeing with the species tree, SINDIR constructed correct trees with a sensitivity of 85% and a specificity of 95%. We have also compared the algorithm's performance on 100 mammalian gene families where Neighbor Joining and Maximum Likelihood result in long branch attraction error, and in each case the correct tree was reconstructed by our method. Our initial results suggest that phylogenetic reconstruction based on a species tree can outperform current methods that ignore species information. SINDIR provides a fast, reliable, and practical methodology for genome comparison and evolution studies. 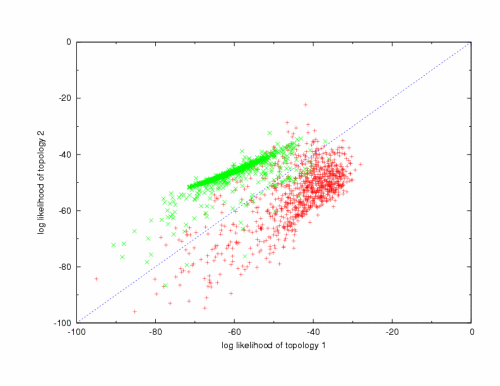 Figure 1: Comparison of two toplogies 1: (dog,(human,(mouse,rat))) and 2: ((dog,human),(mouse,rat)) for 1800 gene clusters. Clusters that are actually topology 1 are ploted red, and clusters of topology 2 are ploted green. References:[1] Maido Remm, Christian Storm, and Erik Sonnhammer. Automatic Clustering of Orthologs and In-paralogs from Pairwise Species Comparisons. In Journal of Molecular Biology 314:1041-1052. 2001 [2] Roman Tatusov, Natalie Fedorova, John Jackson, Aviva Jacobs, Boris Kiryutin, Eugene Koonin, Dmitri Krylov, Raja Mazumder, Sergei Mekhedov, Anastasia Nikolskaya, Sridhar Rao, Sergei Smirnov, Alexander Sverdlov, Sona Vasudevan, Yuri Wolf, Jodie Yin, and Darren Natale. The COG database: an updated version includes eukaryotes. In BMC Bioinformatics. 4:41 September. 2003 [3] Jonathan A. Eisen. Phylogenomics: improving functional predictions for uncharacterized genes by Evolutionary Analysis. In Genome Research. 8(3):163-7. 1998 [4] N. Saitou and M. Nei. The neighbor-joining method: a new method for reconstructing phylogenetic trees. In Molecular Biology Evolution. 4(4):406-425. July 1987. |
|||
|