| Research Abstracts Home | CSAIL Digital Archive | Research Activities | CSAIL Home |
![]()
|
Research
Abstracts - 2007
|
|
Molecular Design of Drug Resistance Resistant InhibitorsDavid J. Huggins, Nate Silver & Bruce TidorIntroductionAIDS is one of the major killers in the modern world, but despite the many treatments that have been developed, the HIV virus continues to spread. One of the main problems in designing drugs to combat HIV is the potential for the virus to develop resistance via mutation. This is a problem that affects drugs targeted at many organisms and is likely to become more of a problem with increased use of drugs to target viral and bacterial infection. We have developed a strategy to help prevent this type of resistance by designing the drug molecule to mimic the exact shape of the substrates of the protein targets. The idea being that any mutations that affect drug binding will also affect substrate binding and thus have a deleterious effect on the organism. This is termed the substrate envelope hypothesis. MethodsThe first step in our drug design scheme is the generation of the substrate envelope, which is created by taking a mould of the targets active site from a crystal structure of the target bound to the substrate. This envelope acts as a barrier, and any designs which pierce this barrier are removed, as they could potentially be knocked out by viral mutants without affecting substrate binding (See Figure 1 ).
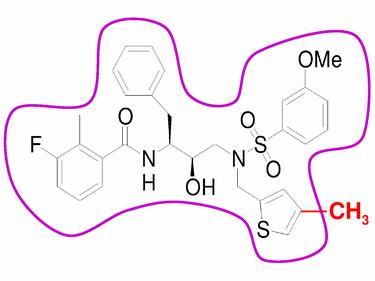
Figure 1 A candidate design which almost fits within the substrate envelope
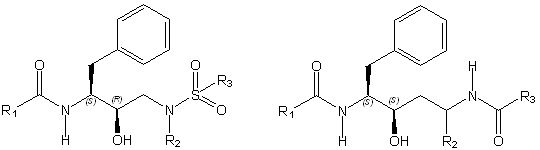
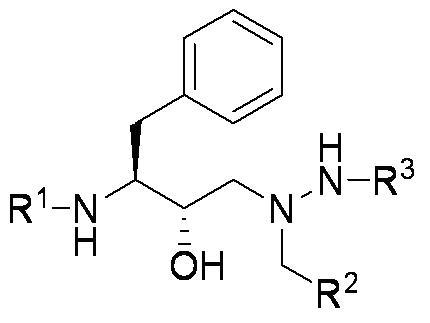
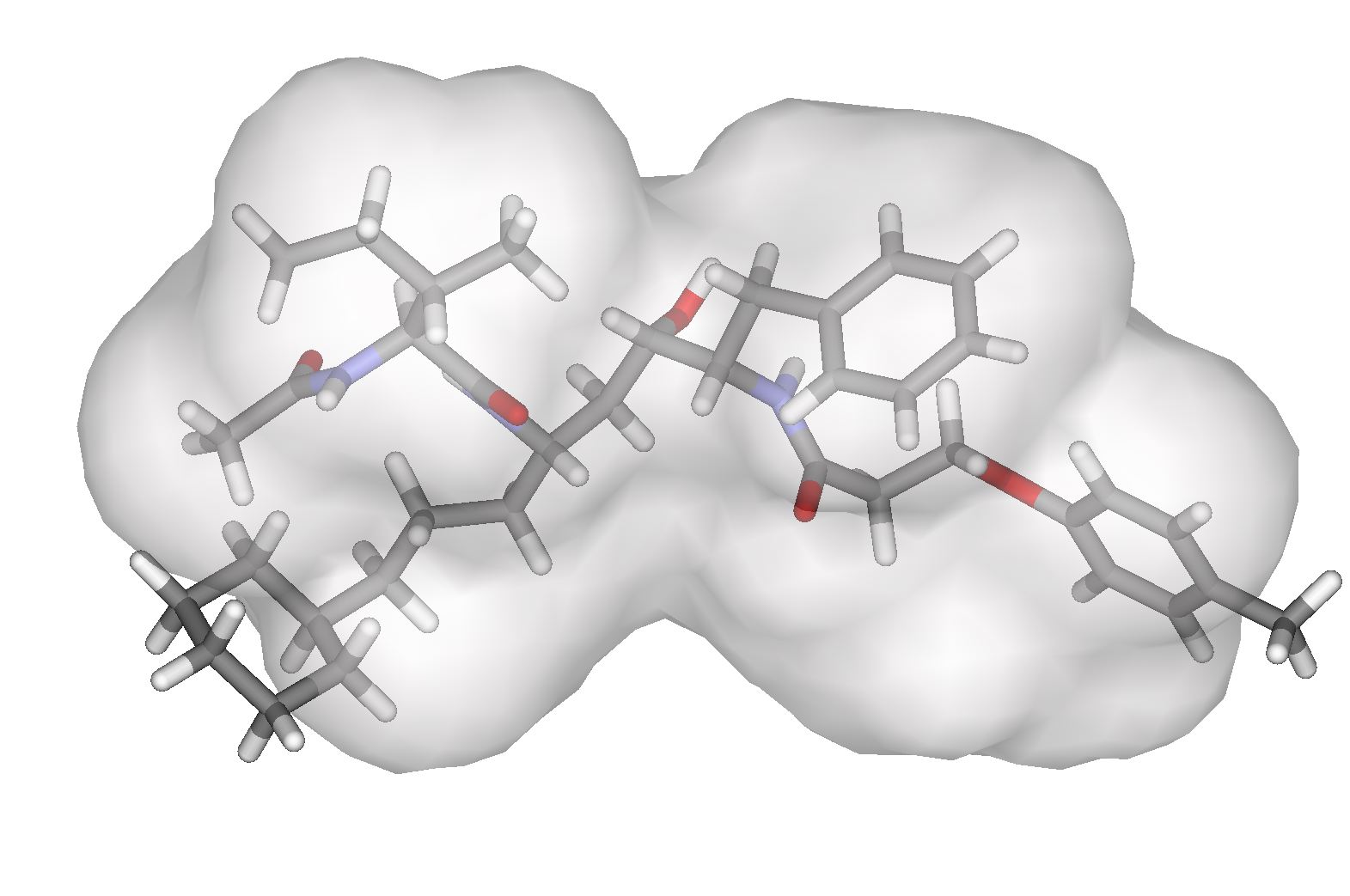
To aid in the synthesis of our designs and reduce the computational load, a fixed scaffold is used as a basis for inhibitor design. These scaffolds are based off of known, successful drugs and are either flexibly or rigidly docked within the envelope. They contain a number of variable sites upon which different functional groups can be grown, and by searching this functional group space, we are able to design inhibitors with a high predicted free energy of binding. However, the large number of scaffold positions combined with a large number of choices at each position and the necessity to search many conformations make this a complex problem. We use a combination of dead end elimination [1] and the A* algorithm [2] to analyze the many possibilities. This allows us to search a large portion of the space in a reasonable amount of time. Model SystemsAll of our work thus far has been focused on designing inhibitors to HIV-1 protease, as it is a widely studied system and is considered to be one of the most effective targets to slow the spread of HIV. We use the backbone structures of three of the most potent HIV inhibitors, Amprenavir, Lopinavir, and Atazanavir (See Figure 2) as our drug scaffolds.
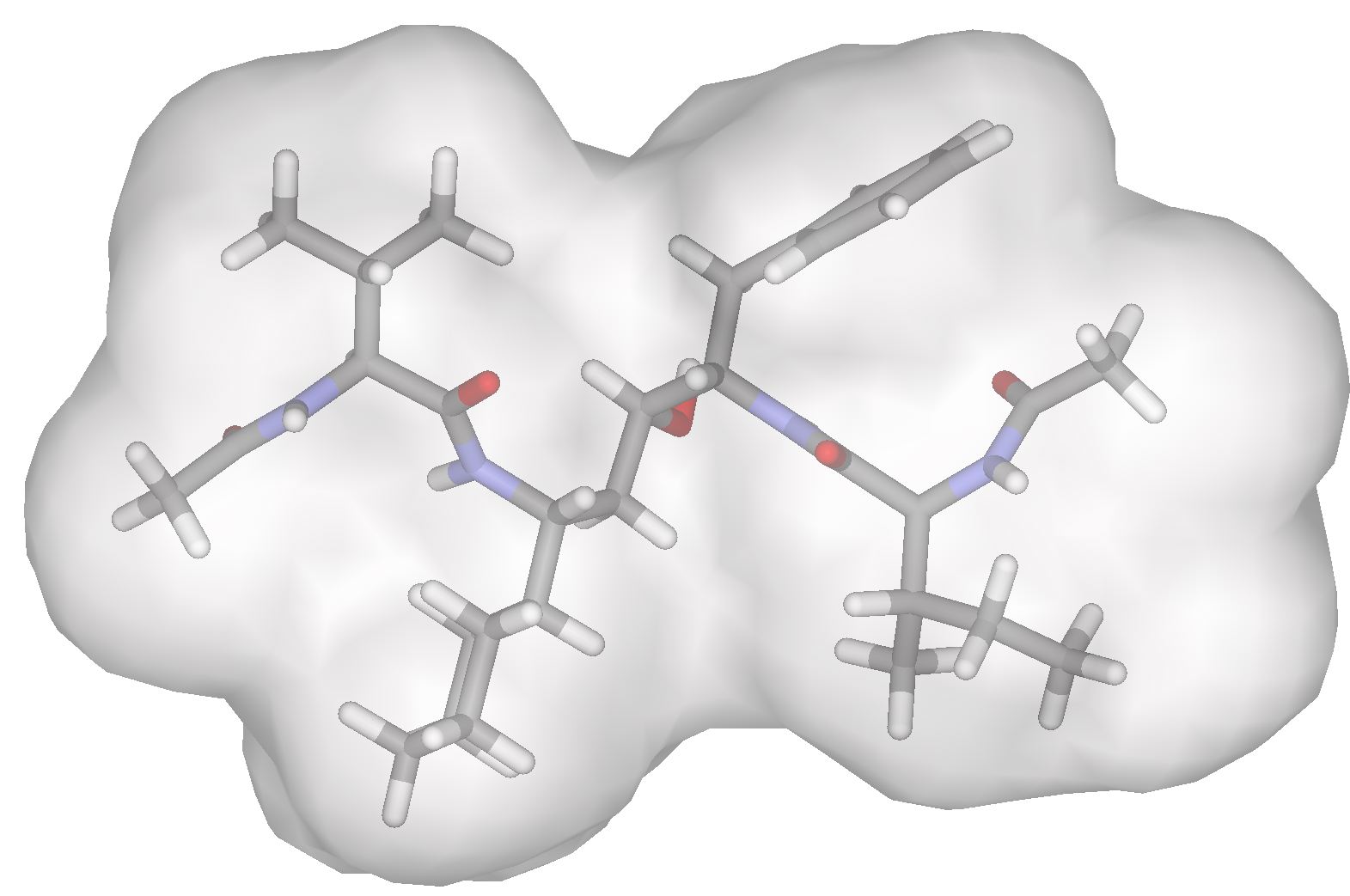
Figure 2 The scaffolds of Amprenavir, Lopinavir and Atazanavir used for inhibitor design. A variety of commercially available compounds are used at the R1, R2 and R3 positions to design inhibitors with a high predicted free energy of binding. Current ProgressA set of compounds based on the core of Amprenaivir have been designed, synthesised and tested against a panel of four HIV-1 Protease mutants that represent the most commonly occurring mutations. Results are promising. The tightest binding compound has an affinity of 14pM by isothermal titration calorimetry and, from the set of compounds that have been tested against the mutant panel, the best lose less than tenfold affinity. We have also designed a series of compounds from the Lopinavir core. There are two sets of compounds. Half fit within the substrate envelope and half are larger and predicted to bind more tightly but do not fit within the substrate envelope. Examples of both of these sets of compounds can be seen in Figure 2. All these compounds are being tested for affinity and against the panel of mutants.
Figure 2 Compounds predicted to bind inside (left) and outside (right) the substrate envelope We have also been working with medicinal chemists in an attempt to optimise some highly promising compounds based on the amprenavir scaffold. The marriage of experiment and theory will hopefully yield some highly promising inhibitors. Future PlansWe hope to prove the concept of the substrate envelope hypothesis by designing and making molecules that mimic the shape of the peptide substrate of HIV protease and have good activity against a variety of mutant proteases. We can then establish a simple and effective methodology for to avoiding the problems associated with drug resistance in the future. This could greatly aid future efforts in drug discovery. AcknowledgementsThe Amprenavir predictions and much of the coding for this work was done by Michael D. Altman. References:[1] J. Desmet, M. De Maeyer, B. Hazes and I. Lasters. The dead-end elimination theorem and its use in protein side-chain positioning. Nature 356, p539542, 1992. [2] A. R. Leach and A. P. Lemon. Exploring the conformational space of protein side chains using dead-end elimination and the A* algorithm. Proteins 33, p227239, 1998. |
|||
|