| Research Abstracts Home | CSAIL Digital Archive | Research Activities | CSAIL Home |
![]()
|
Research
Abstracts - 2007
|
|
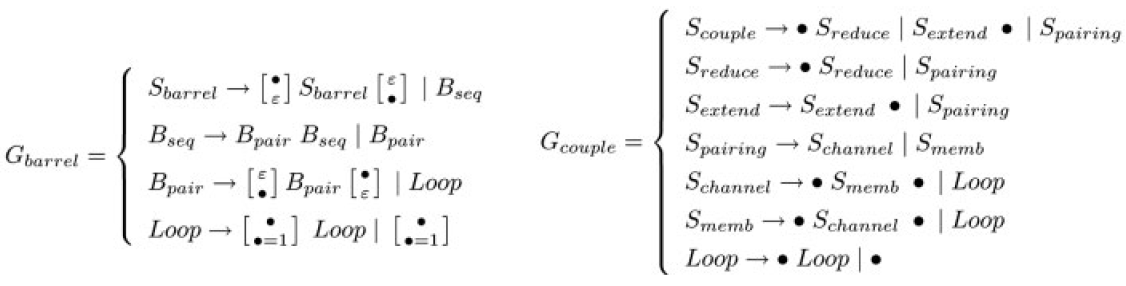
Modeling Ensembles of Transmembrane β-barrel ProteinsJérôme Waldispühl, Charles W. O'Donnell, Nathan Palmer, Srinivas Devadas, Peter Clote & Bonnie BergerThe goal of this project is to develop a whole new family of algorithms for exploring the folding landscape of transmembrane β-barrel proteins. Using the Boltzmann partition function and a bottom-up energy model that does not rely on machine learning, we are able to compute, from sequence, meaningful physical properties and the weighted ensemble of supersecondary structures that a particular TMB might conform to. Current progress on this project can be found at http://protein.csail.mit.edu/ and http://theory.csail.mit.edu/~bab. IntroductionTransmembrane β-barrel (TMB) proteins are embedded in the outer membrane of Gram-negative bacteria, mitochondria and chloroplasts. The cellular location and functional diversity of β-barrel outer membrane proteins (omps) makes them an important protein class. At the present time, very few non-homologous TMB structures have been determined by X-ray diffraction because of the experimental difficulty encountered in crystallizing transmembrane proteins. Our recent algorithm, transFold, is able to compute the minimum energy supersecondary structure for omps, identifying β-strands and their inter-strand H-bond contacts from sequence alone [1]. To do this we use a multitape S-attribute grammar to model a TMB structure, and use dynamic programming to find the minimum energy structure according to statistical potentials of interstrand residue contacts. This approach was the first to predict such long-range interactions for TMBs, and differs significantly from others by not requiring a training phase on known TMB structures. An online webserver has been set up at http://theory.csail.mit.edu/transFold/ where the algorithm can be run on any given sequence [2]. Unfortunately, transFold can only determine minimum folding energy structures, and says nothing about the folding landscape that is being investigated. For the first time, we now move beyond classical single structure prediction methods and introduce a family of algorithms for investigating the folding landscape of TMBs using the Boltzmann partition function. Inspired by previous work on RNA secondary structure, we have developed a recursive algorithm for computing the partition function value, and from this, we are able to compute the individual residue-residue contact probabilities of inter-β-strand contacts. Further, we have devised an algorithm for rigorously sampling structural conformations from the Boltzmann low energy ensemble. By creating stochastic contact maps from the residue-residue contact probabilities, sampling structural conformations according to their probability in the Boltzmann distribution, and computing physical properties from the Boltzmann partition function value, we can gain a much more clear understanding of the entire folding landscape of an outer membrane protein, rather than just the minimum energy conformation. MethodsTransmembrane β-barrel proteins can be modeled using a multitape S-attribute grammar by linearly representing the constituent β-strands by their secondary structure assignment, duplicating this linear assignment to form two tapes, and aligning the two tapes against each other to indicate the alignment of β-strands in a β-sheet, and the location of H-bonds. Using this form, the barrel structure of a TMB (requiring a barrel-shaped β-strand alignment) can be defined by a 2-tape grammar (as shown by Gbarrel and Gcouple rules below). Using this grammar, a parse tree can then be derived, and S-attributes within the tree that correspond to energy potentials can be used to find the optimal supersecondary structure. More details on how to use S-attribute grammars to represent protein structure can be found in [1] and [3].
Inspired by McCaskill's work on RNA structure prediction [4], the Boltzmann partition function can be found for a TMB by projecting the β-barrel structure onto a 2D lattice and solving a dynamic programming recursion. Although this problem is NP-Hard for 3D model, Istrail has shown that a 2D lattice can be solved in polynomial time [5]. In a similar fashion, the individual residue-residue contact probability can be solved according to each contacts weight in the Boltzmann distribution. Finally, independent, random, structural conformations can be sampled from the ensemble of possible conformations, again according to their weight in the Boltzmann distribution. These techniques are inspired by work done by Ding and Lawrence [6]. ResultsUsing our methods [7], a stochastic contact map can be created for any given TMB residue sequence, identifying likely structural conformations in the Boltzmann ensemble. In the figure below, both axis delineate the amino acid sequence, and darker regions in the map represent high-probability interactions according to our derived Boltzmann distribution. The minimum energy structure, as would be found be transFold is also shown in red. Initial results suggest that our residue-residue contact predictions are the best for transmembrane β-barrel proteins when compared to contacts found in x-ray crystal structures.
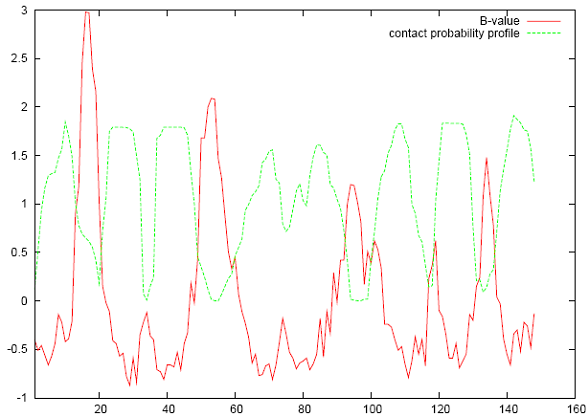
Since residue-residue contact probabilities can indicate regions of a TMB protein that are more flexible, we can compare the per-residue contact probability with the Debye-Waller factor (B-value) found in x-ray crystal structures. As seen in the figure below, sequential residue regions of high flexibility should maintain a low per-residue contact probability (in green). These low-probability regions sequence across the x-axis correspond very well with high experimental B-value regions which also indicate flexible and disordered structure.
References:[1] J. Waldispühl, B. Berger, P. Clote, and J-M. Steyaert. Predicting Transmembrane Beta-barrels and Inter-strand Residue Interactions from Sequence. In PROTEINS: Structure, Function and Bioinformatics, 65(1) pp. 61-74 (2006) [2] J. Waldispühl, B. Berger, P. Clote, and J-M. Steyaert. transFold: a web Server for Predicting the Structure and Residue Contacts of Transmembrane β-barrels. In Nucleic Acids Research, 34 (Web Server issue) pp. W189-W193 (2006) [3] J. Waldispühl and J-M. Steyaert. Modeling and Predicting all-Alpha Transmembrane Proteins Including Helix-Helix Pairing. In Theoretical Computer Science, special issue on Pattern Discovery in the Post Genome, pp. 67-92 (2005) [4] J. S. McCaskill. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. In Biopolymers, 29 pp. 1105-1119 (1990) [5] I. Istrail. Statistical mechanics, three-dimensionality and NP-completeness: I. Universality of intractability of the partition function of the ising model across non-planar lattices. In Proceedings of the 32nd ACM Symposium on the Theory of Computing, pp. 87-96 (2000) [6] Y. Ding, C. E. Lawrence. A statistical sampling algorithm for RNA secondary structure prediction. In Nucleic Acids Research, 31(24) pp. 7280-7301 (2003) [7] J. Waldipühl, C. W. O'Donnell, N. Palmer, S. Devadas, P. Clote, B. Bergeer. Modeling Ensembles of Transmembrane β-barrel proteins. Under review. |
|||||||
|