| Research Abstracts Home | CSAIL Digital Archive | Research Activities | CSAIL Home |
![]()
|
Research
Abstracts - 2007
|
|
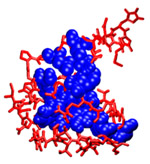
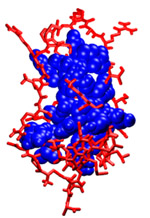
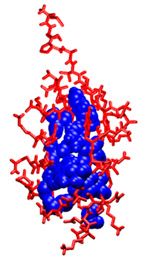
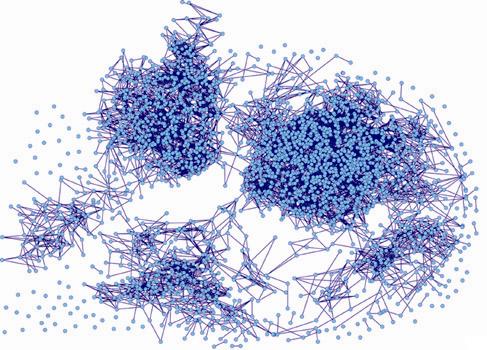
Computationally Mapping Sequence Space to Understand Evolutionary Protein EngineeringKathryn A. Armstrong & Bruce TidorThe design and development of enzymes that extend the catalytic range beyond the currently available repertoire is important for industrial synthesis. Progress in this area has generally involved making use of structural and/or homologous sequence information of a parent enzyme carrying out a related function that is used to seed a search in sequence space. However, the sequence space available for modification during re-engineering is phenomenally large, and such searches can only examine a tiny fraction of it. Protein engineering studies using this class of evolutionary techniques have met with varying degrees of success, making it difficult to draw conclusions about the relative effectiveness of different approaches. It is unclear the extent to which this variability reflects differences in the challenges attempted, in the technology applied, in the sizes or properties of the space of proteins searched, or something about the fundamental adaptability or designability of the parent protein. To explore these issues we computationally examine both the number of sequences allowed for a given structure and the accessibility of these sequences by different protein engineering techniques. We computationally explore the allowed sequence space for small proteins, allowing us to design, simulate, and evaluate strategies for protein design via directed evolution. The core residues for three of these proteins, highlighted in Figure 1, are analyzed using the dead-end elimination and A* algorithms to produce a rank-ordered list of sequences which are compatible with the backbone fold. Some sequences are stable for a given protein backbone structure while others are not, meaning that depending on the search and screening strategies, entire regions of sequence space may be inaccessible. Using computational simulations of error-prone PCR experiments, we then explore these sequence spaces using different experimental strategies. For example, evolutionary design procedures that make mostly single DNA mutations will sample the sequence space nearby the wild-type sequence very well. A new useful protein might be selected from a library of these nearby sequences, if they are allowed for the given protein structure. However, if the goal function requires a new sequence with multiple cooperative mutations, such a method is unlikely to reach that sequence before it generates more sequences than can be screened experimentally. Techniques that sample sequence space more broadly have been useful for finding new proteins with multiple, cooperative mutations. Genetic recombination, focused mutagenesis, and computational design techniques have all been successful in finding design solutions more distant from the wild-type sequence. Our computational approach allows us to explore these experimental strategies in detail. Structure-based protein design allows us to observe how the constraints of different backbone conformations influence the number and type of allowed protein sequences. We make "sequence-space graphs" of these allowed sequences showing the relationships between them in terms of mutation distance and find that for some proteins the allowed sequences are close together in mutational space and in others more separated clusters of sequences are prevalent. These graph topologies give us insight into the most likely evolutionary paths through sequence space; an example sequence-space graph is shown in Figure 2. By testing different error-prone PCR strategies we find that using our sequence-space graphs to guide the choice of wild-type starting sequence can help generate more diverse libraries for screening. The techniques developed for this problem could also be used as a testing platform for other new ideas for evolutionary protein engineering in the future.
|
|||||||||||
|
||||||||||||