| Research Abstracts Home | CSAIL Digital Archive | Research Activities | CSAIL Home |
![]()
|
Research
Abstracts - 2007
|
|
Designing Drugs and Drug Cocktails that Recognize Multiple Targets: Theory and Application to HIV-1 ProteaseMala L. Radhakrishnan & Bruce TidorMotivationA major concern in retroviral treatment is the mutation of the target proteins, resulting in drug resistance over time. This problem has especially hindered HIV treatment, where newly infected patients are already harboring drug-resistant viral mutants, suggesting that such forms are surviving and being successfully transmitted [1]. One currently promising approach is to combine drug molecules into a cocktail such that each molecule recognizes a subset of the target mutants, and together they recognize all variants. Cocktail therapy, such as Highly Active Anti-Retroviral Therapy (HAART) for HIV [2], has been quite successful. Nevertheless, rational cocktail design is still a new prospect, and a general understanding of the determinants of potent cocktails would be useful for future, improved cocktails toward HIV and other diseases. Our ApproachOur goal is to understand general principles that could be of use in rational cocktail design. Ideally, one would like to have a cocktail containing the fewest number of molecules possible yet still accounting for all relevant target mutants. Such a cocktail should contain drugs that are each responsible for covering different target sets, to avoid redundancy. We wish to understand whether it is possible to group targets together within an ensemble by their propensity to be bound by a single drug within the cocktail. To help answer such questions, we are developing integer-programming-based methods to design optimal drug cocktails toward ensembles of targets. These methods select or design the fewest number of drug molecules such that each target is bound by at least one drug molecule with high affinity. We are applying these methods to design optimal cocktails toward theoretical model molecule ensembles; this has allowed us to extract general principles about the relationship between targets in the ensemble and the optimal drug cocktail. Using a physics-based framework grounded in continuum electrostatics [3,4], we are also extracting general principles for understanding physical characteristics that help a molecule recognize multiple targets. The physical properties include molecular charge distribution, conformational flexibility, and molecular size. Modulating such properties to increase broad binding toward targets may be useful in designing the molecular components of a cocktail.
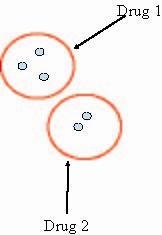
Figure 1. Schematic representation of a two-drug cocktail that covers five targets. One goal is to understand how to properly group the targets such that the minimal number of drugs is required. The physical similarities between targets will likely be a useful metric in grouping them. We are also applying our methods to design cocktails toward ensembles of HIV-protease variants. HIV-protease is a major drug target in HIV therapy that rapidly evades drug binding through evolution. First, we are computationally designing individual drug molecules toward each target. The design procedure involves searching a combinatorially large search space made up of a molecular scaffold and several chemical fragments that could be placed upon the scaffold to produce complete molecules. Physics-based energy functions are used to compute binding energies and the global optimization over the large search space is greatly simplified with the Dead-End Elimination algorithm [5,6]. Currently, our target ensemble includes seven HIV-protease variants. Through analyzing the results of the individual designs and also through designing optimal cocktails using our integer-programming methods, we are beginning to uncover specific physical characteristics of molecules in this system that are especially "promiscuous," such that they bind to many of the variants.
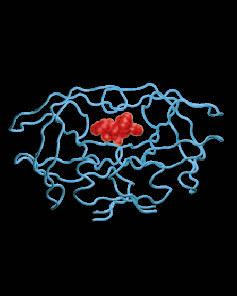
Figure 2. HIV-1 protease structure bound to amprenavir, one of many clinically-approved inhibitors (PDB ID 1HPV) [7]. There are now several drug-resistant variants of the protease, all of which should be considered in effective therapy. Overall, this work will provide useful methods in molecular design toward multiple targets, as well as a useful framing of the cocktail design problem for future applications. Acknowledgements and Research SupportThe authors acknowledge Michael Altman for developing the drug-design software used in this work. This work is funded by the National Institutes of Health and by a Department of Energy Computational Science Gradaute Fellowship (DE-FG02-97ER25308). References:[1] S. J. Little et al. Antiretroviral-Drug Resistance Among Patients Recently Infected with HIV. New Eng. J. Med. 347:385--394, 2002. [2] P. Bonfanti, A. Capetti and G. Rizzardini. HIV Disease Treatment in the Era of HAART. Biomed. and Pharmacotherapy 53:93--105,1999. [3] L.--P. Lee and B. Tidor. Optimization of electrostatic binding free energy. J. Chem. Phys. 106:8681--8690, 1997. [4] E. Kangas and B. Tidor. Optimizing Electrostatic Affinity in Ligand--Receptor Binding: Theory, Computation, and Ligand Properties. J. Chem. Phys. 112:9120--9231, 2000. [5] J. Desmet, M. D. De Maeyer, B. Hazes, and I. Lasters. The Dead-End Elimination Theorem and its use in Protein Side-Chain Positioning. Nature 356:539--542, 1992. [6] R. F. Goldstein. Efficient Rotamer Elimination Applied to Protein Side-Chains and Related Spin Glasses. Biophys. J. 66:1335-1340, 1994. [7] E. E. Kim et al. Crystal Structure of HIV-1 Protease in Complex with VX-478, a Potent and Orally Bioavailable Inhibitor of the Enzyme.J. Am. Chem. Soc. 117:1181--1182, 1995. |
|||
|