| Research Abstracts Home | CSAIL Digital Archive | Research Activities | CSAIL Home |
![]()
|
Research
Abstracts - 2007
|
|
Information Flow Analysis of Interactomes to Corroborate Phenotypic Knockout Profiles of GenesPatrycja V. Missiuro & Hui Ge*MotivationThe goal of the presented work is to understand how analysis of protein interaction networks can corroborate and predict loss-of-function phenotypesof genes or gene pairs. We need to be able to effectively model relevant local and global properties of biological networks taking into account noise and variable data quality, and to be able to dynamically incorporate new data as it becomes available. To achieve that, we have developed information flow metric that we use to find globally important genes which have not been detected with previous approaches. In addition, we successfully combine information flow with previously tried metrics to detect more phenotype-enriched genes. BackgroundAdvances in biological experimentation methods have enabled large scale studies of protein-protein interactions in many species. Much of the high throughput data is freely available, and we can combine it to generate interactomes. Interactomes are networks where each protein is modeled as a node and its interactions with other proteins as either binary or weighted undirected edges. Given a network representation of the specie’s proteome, one can perform local and global network analyses of individual or groups of proteins. The resulting metrics can then be compared with other types of biological data to determine whether one is predictive of another. For example, embryonic lethal phenotype has been successfully linked to high degree nodes, those with high number of interacting partners [1]. A global measure of betweenness has been used to describe node’s centrality in the network by counting how many shortest paths run through it [2]. However, correlating betweenness with phenotypes has not been successful and we hypothesize it is because it considers only the shortest paths. Current methods to analyze properties of networks, among those degree and betweenness, are designed to only effectively handle binary interaction data. However, with increasing number of available datasets, it is now possible to estimate the probability of interaction between proteins and represent it with a corresponding edge weight.
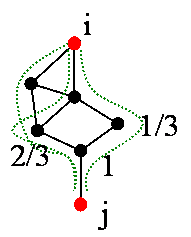
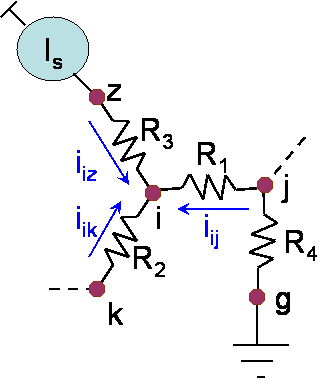
Figure 1: Network metrics of degree, betweenness, and information flow. Betweenness is a fraction of shortest paths running through a node. Information flow takes into account relative contribution of all possible paths. ApproachWe introduce information flow, a novel network analysis method, and use it to identify lethal or phenotypic genes based on the flow estimates of their network importance. Information flow method takes into account edge weights and automatically considers all possible paths. Given that currently available interactomes are noisy and incomplete, this allows for a consistent and more globally accurate estimate of gene’s significance as the network grows and becomes more dense when more data becomes available. Information flow method works with interactome modeled as a resistor circuit (see figure 2). Resistor values are inversely proportional to protein interaction probabilities. Information flow through a protein node is an absolute sum of current flow through that node as we iterate over all combinations of remaining node pairs, assigning one as a current source and second one as a sink node. Our method is exact and computationally efficient, and it takes into account all possible paths, weighting them by their likelihood. . We can detect nodes which are central to the network as well as those which belong to slightly weaker alternative pathways, which would have otherwise remained undetected.
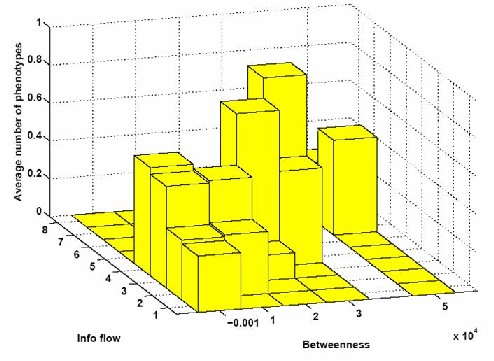
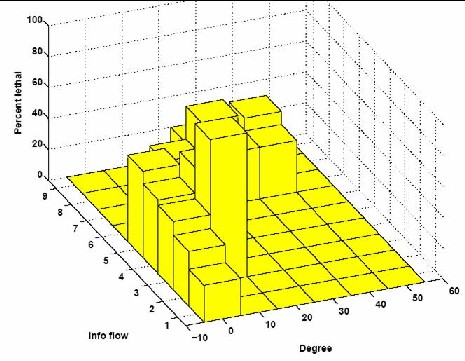
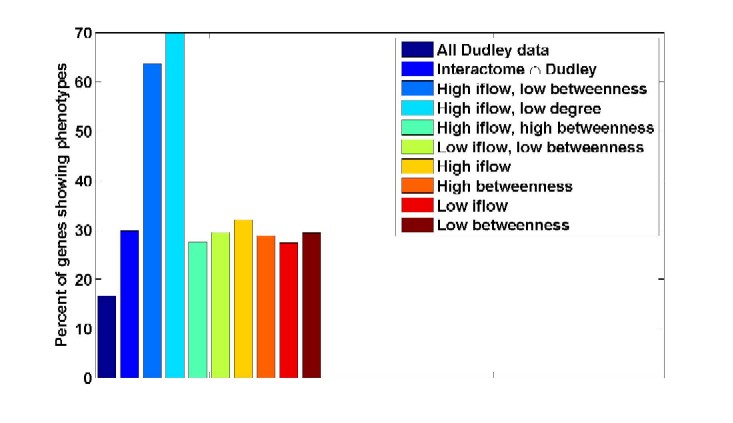
ResultsWe have promising results of our network analysis when applied to two different interactomes, C. elegans (worm) [3, 4] and yeast [5], where the edges are binary and weighted, respectively. We show how information flow, unlike betweenness, can predict subtle gene properties such as presence and number of phenotypes (figure 3a). When combined with degree, it can pinpoint lethal nodes which would not be detected by degree alone (figure 3b). When combined with either degree or betweenness, it predicts otherwise undetected pleiotropic genes (see figure 4).
Figure 3: C. elegans interactome statistics Future directionsWe hypothesize that combining information flow metric with others can be used to pinpoint alternative interaction pathways that may correspond to new and more subtle regulatory processes. We plan to use information flow scores along with results from studying other types of high-throughput data as features useful for modeling various biological processes.
FIGURE 4: Percent of genes exhibiting phenotypes vs their network statistics. Research supportFinancial support from the Whitehead Institute is gratefully acknowledged. References[1] H. Jeong, S.P. Mason, A. L. Barabasi and Z. N. Oltvai, Lethality and centrality in protein networks, Nature 411, 41, 2001. [2] Freeman, L. C., A set of measures of centrality based on betweenness, Sociometry 40, 35--41, 1977 [3] Source of data: http://www.wormbase.org [4] S. Li, C. M. Armstrong, N. Bertin, H. Ge, M. Vidal et al., A map of the interactome network of the metazoan C. elegans, Science 303, 540-543, 2004 [5] Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dümpelfeld B, et al., Proteome survey reveals modularity of the yeast cell machinery, Nature, 440:631-636, 2006 [6] M. Girvan and M. E. J. Newman, Community structure in social and biological networks, in Proceedings of National Academy of Sciences USA, Vol. 99, No. 12., pp. 7821-7826, 11 June 2002 *Hui Ge is a Fellow at the Whitehead Institute of Biomedical Research. |
||||||||
|