
| Research Abstracts Home | CSAIL Digital Archive | Research Activities | CSAIL Home |
![]()
|
Research
Abstracts - 2007
|
|
Roles of Gene & Species Mutation Rates for Accurate Gene PhylogeniesMatthew D. Rasmussen & Manolis KellisAbstractComparative genomics provides a general methodology for discovering functional DNA elements, and understanding their evolution [1-4]. Comparisons of many genomes can be more powerful, but require rigorous phylogenetic methods to resolve orthologous genes and regions. Here, we address the problem of accurate gene tree reconstruction across many complete genomes, using twelve Drosophila and nine Saccharomycete species. We show that existing phylogenetic methods which treat each gene tree in isolation show large-scale inaccuracies, largely due to insufficient phylogenetic information in individual genes. However, we find that gene trees exhibit common properties, which can be exploited for accurate phylogenetic reconstruction. Evolutionary rates can be decoupled into gene-specific and species-specific components, which can be learned across complete genomes. We develop a maximum-likelihood methodology for phylogenetic reconstruction which exploits these properties, and show that it achieves significantly higher accuracy, addressing the long-branch-attraction problem, and enabling studies of gene evolution in the context of species evolution. Gene & species trees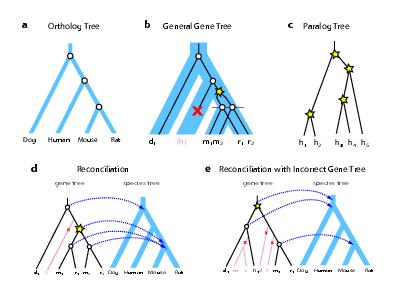 Relationship between gene trees and species trees. a-c. Ortholog trees imply species relationships (a), and paralog trees imply gene family expansions within a single species (c). General gene trees (b) combine both orthologs and paralogs across multiple species to infer gene duplication (star), gene loss (x), and speciation (circle). Each gene is named with the first letter of the corresponding species The gene tree (black lines) can be viewed as evolving inside the species tree (blue area), implying coordinated speciation events at branching points in the species tree (dotted line). d. Gene duplication and loss events are inferred by reconciling a gene tree to a species tree, mapping each gene-tree node to its closest species-tree common ancestor node (arrows). e. When the gene tree is incorrect, many spurious events will be inferred. In this example, a common misplacement of rodents due to long-branch-attraction leads to four spurious events (one duplication and at least three losses). References:[1] Mouse Genome Sequencing Consortium. Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520-562 (2002).
[2] Kellis, M., Patterson, N., Endrizzi, M., Birren, B. & Lander, E. S. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423, 241-254 (2003). [3] Richards, S. et al. Comparative genome sequencing of Drosophila pseudoobscura: chromosomal, gene, and cis-element evolution. Genome Res 15, 1-18 (2005). [4] Xie, X. et al. Systematic discovery of regulatory motifs in human promoters and 3' UTRs by comparison of several mammals. Nature 434, 338-345 (2005). |
|||
|