| Research Abstracts Home | CSAIL Digital Archive | Research Activities | CSAIL Home |
![]()
|
Research
Abstracts - 2007
|
|
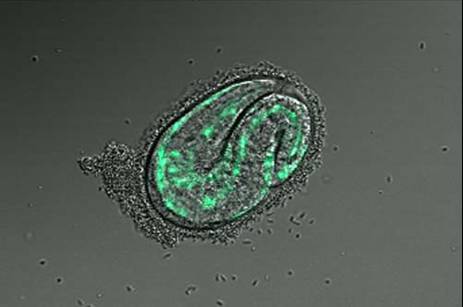
Identifying Tissue-Specific Genes with Semi-supervised LearningPatrycja V. Missiuro & Hui Ge*MotivationGene expression pattern data describes spatial localization of genes in tissues such as muscle, intestine, hypodermis, neurons etc. Knowing where the gene is expressed can help in understanding its function and suggest possible physical, regulatory, or genetic interactions it may be involved in. Recent advances in biological techniques enabled large scale studies of genes and their spatial expression patterns, however, the nature of high throughput experiments is such that the results are noisy. Our objective is to provide a measure of confidence for spatial localization of genes in high throughput datasets. In addition, we would like to identify candidate genes enriched in various tissues based on their similarities to well-known tissue specific genes. BackgroundResults from biological experiments can be split into two categories: high quality but low coverage and those which are noisy but cover a large percentage of genes. Promoter GFP::Fusion method is the former type, because each gene is processed and eye screened individually. In the GFP::Fusion experiment, a promoter targeting a single gene of interest is fused with green fluorescent protein, and genes are visually localized by their green staining in specific tissues [1] (figure 1).
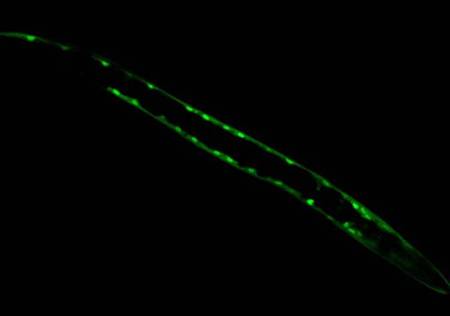
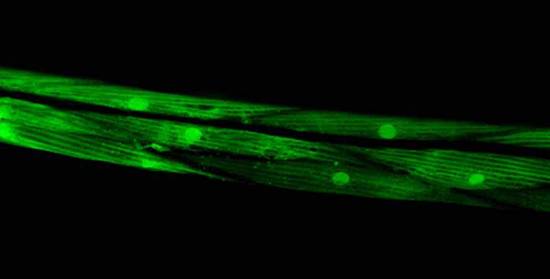
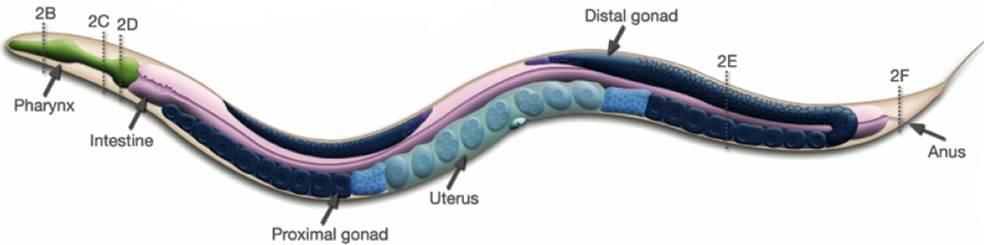
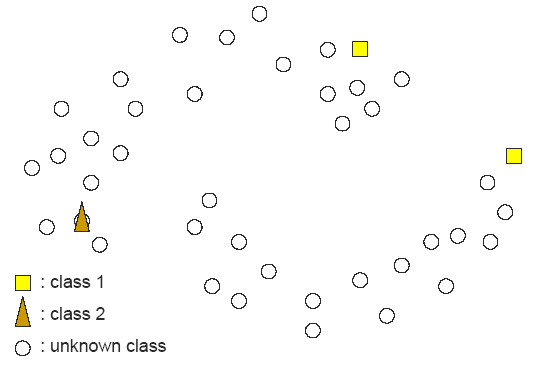
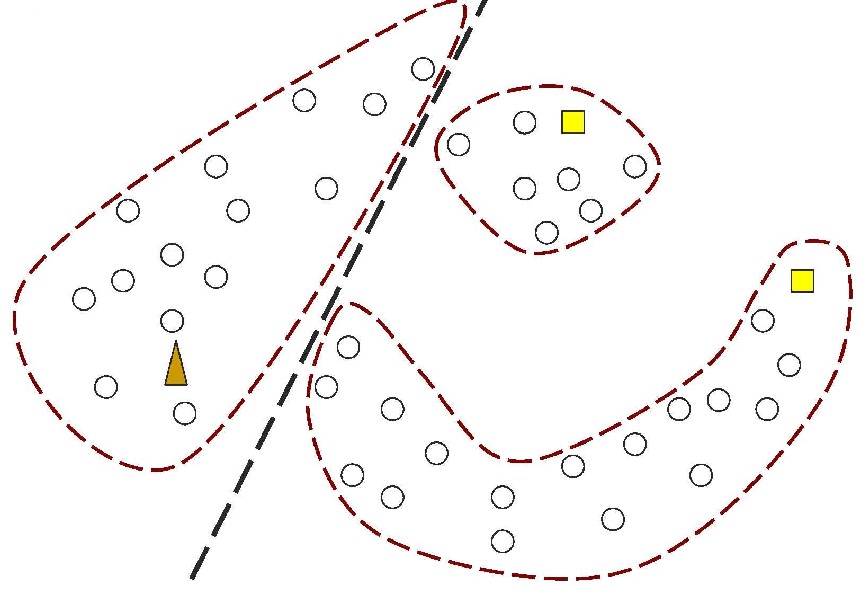
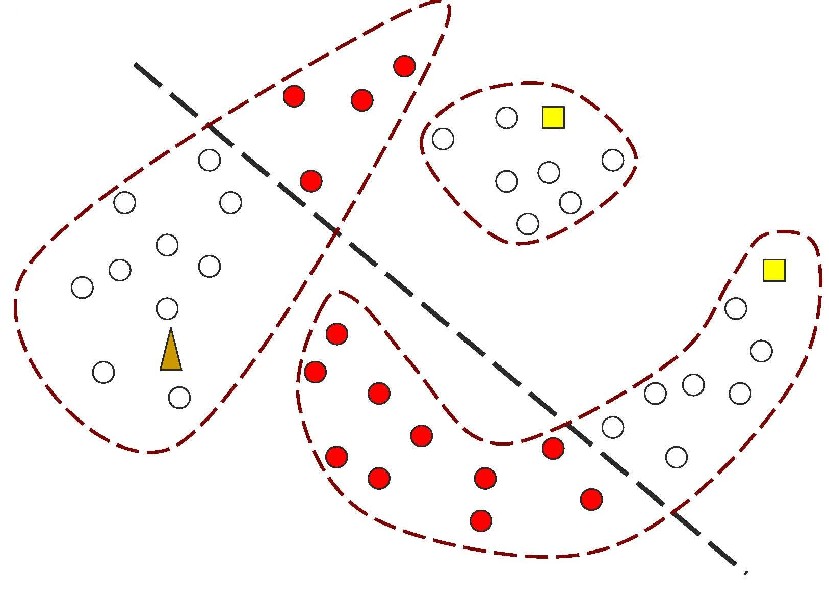
Figure 1: In contrast, Serial Analysis Gene Expression (SAGE) [2] is a high throughput experimental technique where large fraction of genome is simultaneously analyzed. The underlying assumption is that a 14-base pair segment of DNA, called a SAGE tag, from a defined location in each expressed gene, is a unique identifier for a particular gene. Thousands of SAGE tags are simultaneously extracted from a specific tissue to get quantitative analysis of relative levels of the genes expressed. SAGE experiments have been identified as noisy - sources of noise include incorrect tag to gene mappings for tissues, single tag matching multiple genes, and missed gene data [3]. Our model organism is Caenorhabditis elegans, which is a small (1 mm long) soil nematode (worm) (figure 2). C. elegans is one of the most studied organisms thanks to its small size, transparent body, fast development cycle, and presence of many major genes and biological pathways similar to mammals. Transparent worm embryos are ideal for observation of reporter gene expression and they can be manipulated in large quantities, a helpful factor for high-throughput studies. SAGE data currently available covers approximately 10,000 genes which is more than half of C. elegans genome. We also performed a literature search to obtain a list of high quality tissue specific genes coming from Promoter GFP::Fusion and other high confidence, low throughput studies. Figure 2: Tissues in C. elegans hemaphrodite (Source: wormatlas.org) ApproachTo analyze and predict spatial localization of genes we use a novel semi-supervised classification method, Bayesian GENeralization from examples (BGEN) [4, 5]. We use cross validation performance and ROC to compare its classification accuracy with a fully supervised method of Support Vector Machines [6]. The difference between BGEN and SVM is the fact that BGEN uses information from both labeled and unlabeled data, while SVM relies only on the features of the labeled data. BGEN uses information from the overall distribution of feature vectors in all genes when constructing a graph-based prior, which effectively puts a bias for genes to belong to the same class if their feature vectors are similar. Next, the prior distribution on gene classifier is combined via Bayes rule with likelihood function for genes to have a particular label based on labeled data. The resulting posterior distribution is used to classify the entire genome. This way, BGEN biases the classifier not to split clusters of genes with similar phenotypes. In contrast, SVM picks the classification boundary based solely on the labeled data. Figure 3 shows a toy example on how the results from two methods may differ, especially when the training set is relatively small (as in our case).
ResultsFor input, we have a twelve-feature vector per gene, where each feature describes the quantity of individual gene’s tags found in a particular SAGE tissue (twelve tissues) as shown in figure 4.
Figure 4: Example of SAGE feature data. For training input, we hand-curate from literature sets of tissue specific genes (positive control) and genes not present in a specific tissue (negative control). We iterate over all tissues of interest and prepare a distinct training set specific for a given tissue. The output is a probability score which represents the likelihood of a particular gene to be present in a specific tissue. We randomly select muscle tissue as an example. Muscle training set curated from the literature contains 28 well-studied muscle enriched genes and 544 negative controls. SAGE dataset has 9042 unique genes. Since SAGE data contains many genes which have mostly zeros in the tag counts, we threshold the SAGE data and remove any genes where the total sum of tags across tissues is less than 4. The results of thresholding and intersection with training set are summarized in Table 1.
Table 1: SAGE counts before and after intersecting with the training data. Cross validation results from training BGEN and SVM on SAGE data to find muscle-enriched genes show that BGEN has 20% larger ROC area than SVM as shown in figure 5. We run BGEN and predict 7% of unlabeled genes to be muscle enriched, selecting those with likelihood score greater than 0.5. 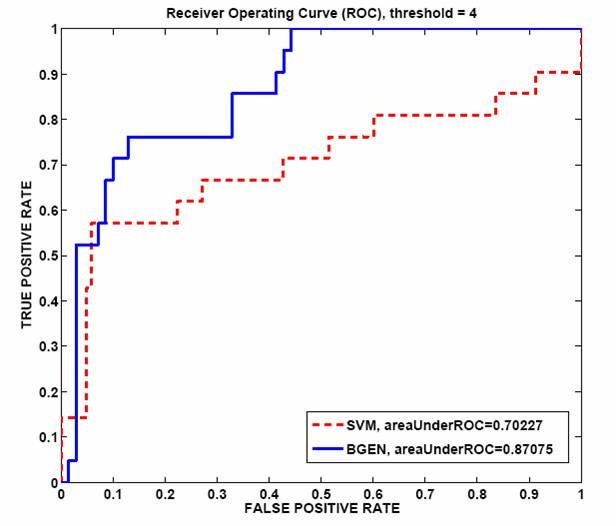 Figure 5: ROC curve comparing BGEN and SVM classifiers for muscle data. Future directionsWe plan to test a random subset of predicted tissue enriched genes at our bio lab at the Whitehead Institute by Promoter GFP::Fusion experiments. Moreover, we are in the process of making predictions for other tissues. Once more tissues are classified, we will integrate our results with other gene features which can help us in solving other biological questions. Research supportFinancial support from the Whitehead Institute is gratefully acknowledged. References[1] Baillie Lab, GFP::Promoter Spatial Expression data, Simon Fraser University, Burnaby, B.C. Canada [2] Velculescu, V. E., Zhang, L., Vogelstein, B., and Kinzler, K. W. Serial analysis of gene expression. Science, 270, 484-487, 1995 [3] Stollberg, J., Urschitz, J., Urban, Z. and Boyd, C.D. Stollberg et al, A quantitative evaluation of SAGE, Genome Research, 10, 1241{1248}, 2000. [4] Yuan (Alan) Qi, Patrycja E. Missiuro, Ashish Kapoor, Craig P. Hunter, Tommi S. Jaakkola, David K. Gifford and Hui Ge, Semi-supervised analysis of gene expression profiles for lineage-specific development in the Caenorhabditis Elegans embryo, Bioinformatics, vol. 22, no. 14, e417-e423, 2006. [5] Thomas Minka, A family of algorithms for approximate Bayesian inference, MIT PhD thesis, 2001. [6] Vladimir Vapnik, Statistical Learning Theory, Wiley-Interscience, New York, 1998. [7] Peter J. Roy, Joshua M. Stuart, Jim Lund, Stuart K. Kim, Chromosomal clustering of muscle-expressed genes in C. elegans, Nature, 418, 975-9, 2002. *Hui Ge is a Fellow at the Whitehead Institute of Biomedical Research. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|